VASP计算Si的Cubic diamond结构理解第一性计算的能带
之前在看第一性计算的文章时,总会看到对于哪个能带,它的主要贡献是来自于哪个原子的哪个轨道的贡献,一直对这一点都是很不解,因子自己的研究方向涉及到拓扑,那么经常也会接触到能带反转,这个时候区分哪个轨道对哪个能带产生贡献,分析是否发生能带反转就变的特别重要,最近正好在参考官网上面的实例学习vasp简单的计算,也接触到了这方面的内容,正好对这个内容进行一下整理,也整理一些自己的理解.
{:.info}
Cubic diamond Si计算
POSCAR
1 | cubic diamond # 体系命名 |
INCAR
1 | System = diamond Si |
KPOINTS
1 | k-points |
有了上面的文件之后,再从赝势库里面找到Si原子的赝势,命名为POTCAR和上面的文件放在相同的文件夹内,然后运行vasp
mpirun -np 20 vasp
上面的计算执行完成之后,会产生一个CHGCAR的文件,利用这个文件就可以进行接下来的计算.
态密度(DOS)
INCAR
接下来就是利用上面计算好的输出文件,进行进一步的性质计算,首先需要对INCAR文件进行修改1
2
3
4
5
6System = diamond Si
ISTART = 0
ICHARG = 2
ENCUT = 240
ISMEAR = -5 # 另外一种计算能带占据函数的方法
LORBIT = 11 # 决定 PROCAR or PROOUT 的文件读写
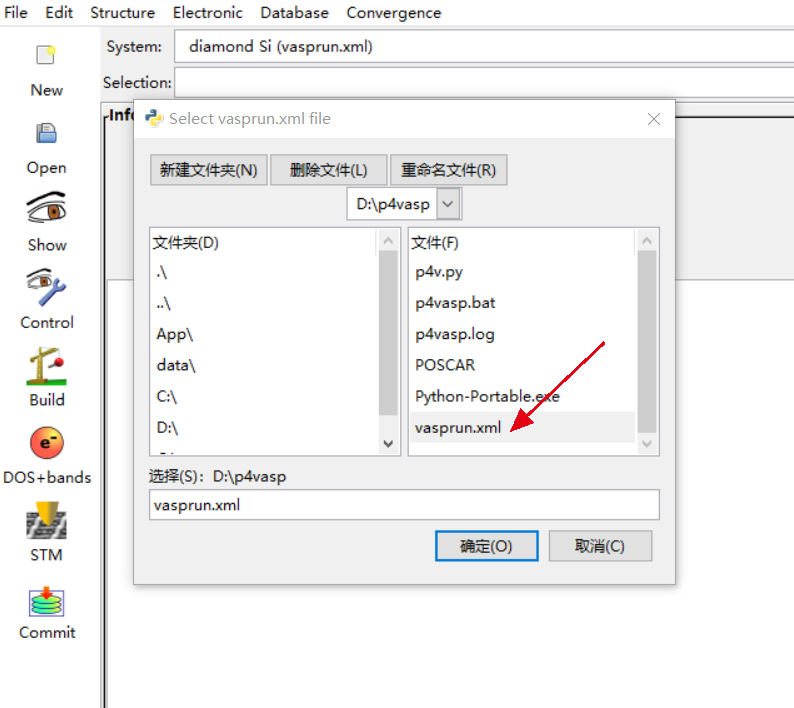
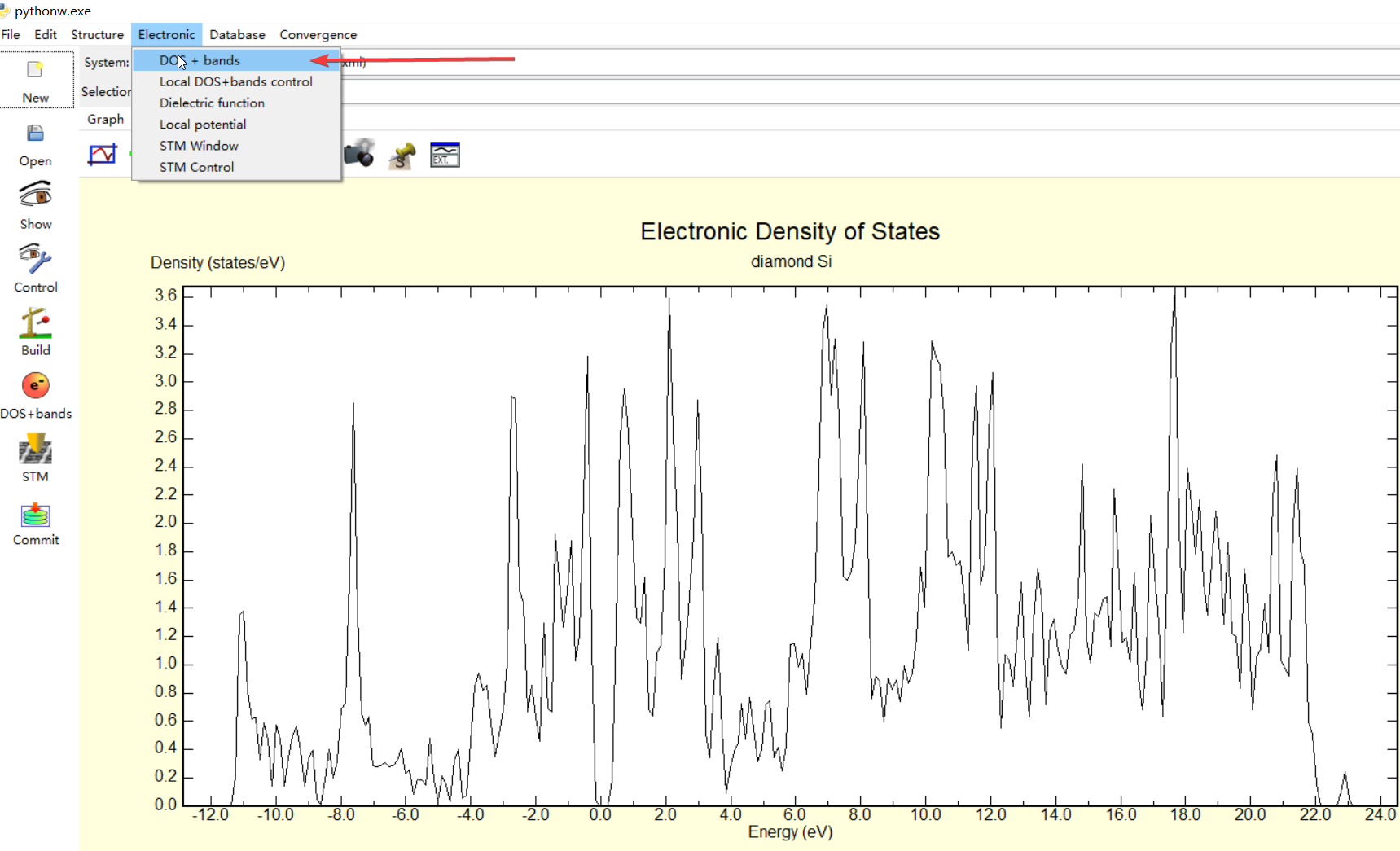
修改INCAR文件之后,再继续运行一次vasp,就可以得到想要的DOS了,不过这个作图需要借助p4vasp.首先计算完成后会有一个vasprun.xml的文件,这就是p4vasp所需要的文件
首先利用p4vasp打开这个文件,然后选择$Electronic\rightarrow Dos+bands$,就可以得到DOS的图
能带计算
在这里同样是首先对INCAR文件进行修改1
2
3
4
5
6
7System = diamond Si
ISTART = 0
ICHARG = 11 # 需要利用前面计算得到的电荷密度文件来对能带进行计算
ENCUT = 240
ISMEAR = 0
SIGMA = 0.1;
LORBIT = 11 # 决定一些文件是否会进行读写
因为要计算能带,那么必然是要沿着一定的高对称线计算,所以此时KPOINTS文件也同样需要修改1
2
3
4
5
6
7
8
9
10
11
12
13
14
15kpoints for bandstructure L-G-X-U K-G
10 # k点数目
line # 能带计算模式选择
reciprocal # 倒空间中k点坐标
0.50000 0.50000 0.50000 1
0.00000 0.00000 0.00000 1
0.00000 0.00000 0.00000 1
0.00000 0.50000 0.50000 1
0.00000 0.50000 0.50000 1
0.25000 0.62500 0.62500 1
0.37500 0.7500 0.37500 1
0.00000 0.00000 0.00000 1
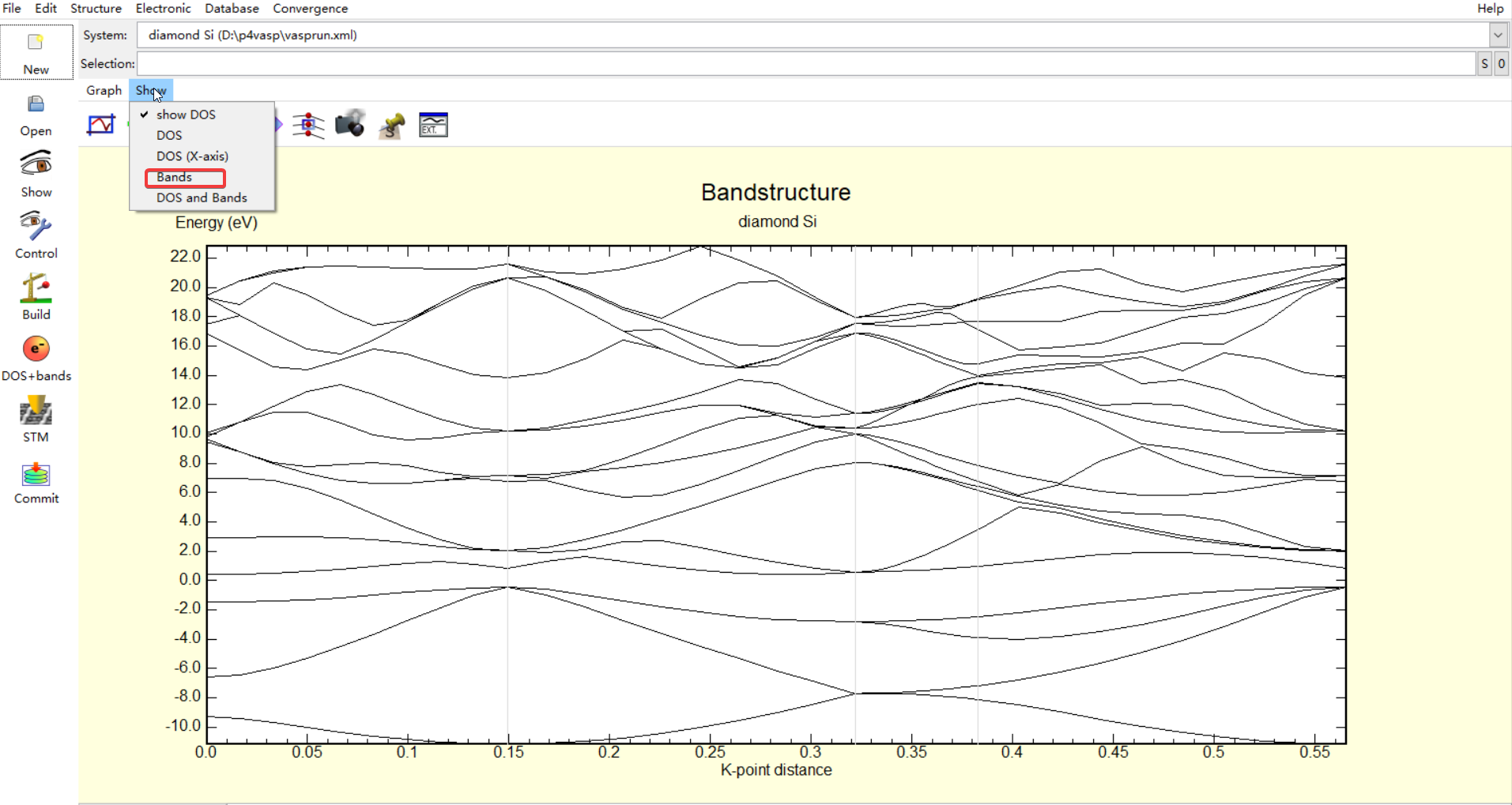
vasprun.xml文件的打开操作和DOS中的操作时相同的,这里有一些需要进行调整,首先要调整显示为band,如下图所示
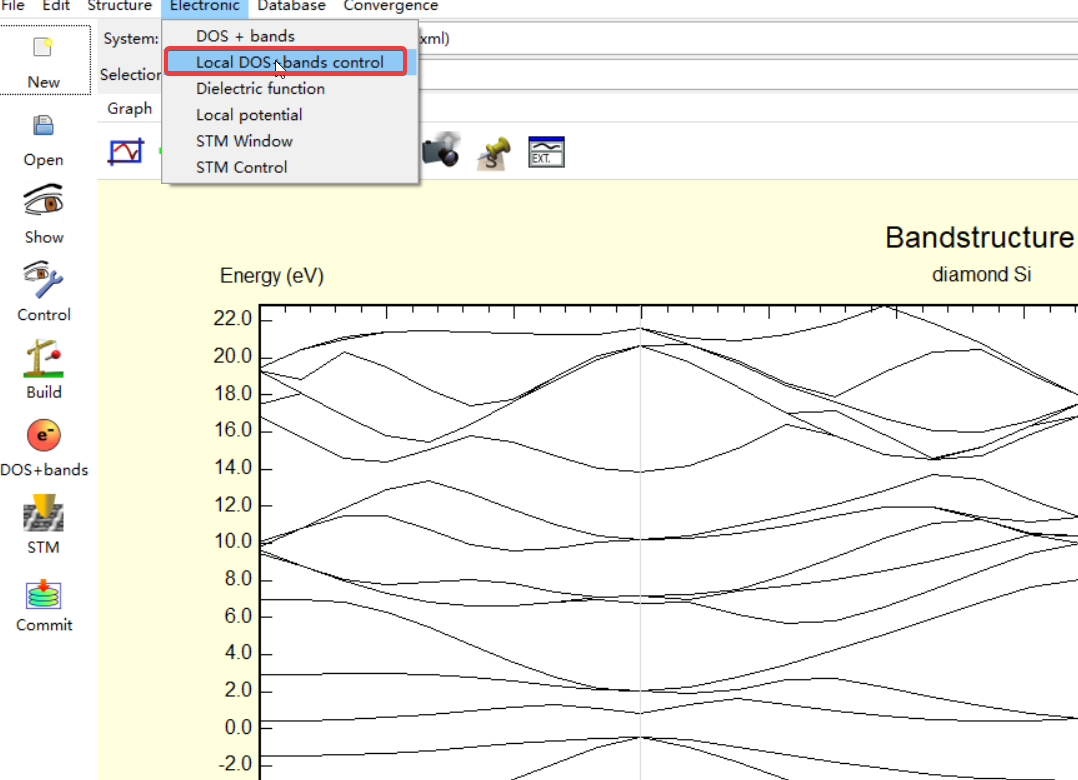
下面就可以分别对不同轨道的贡献进行标记,因为这里只有Si原子,所以也就不存在不同原子不同轨道对能带贡献的说法,不过如果计算的体系包含多种不同的原子,下面的操作同样也可以进行.
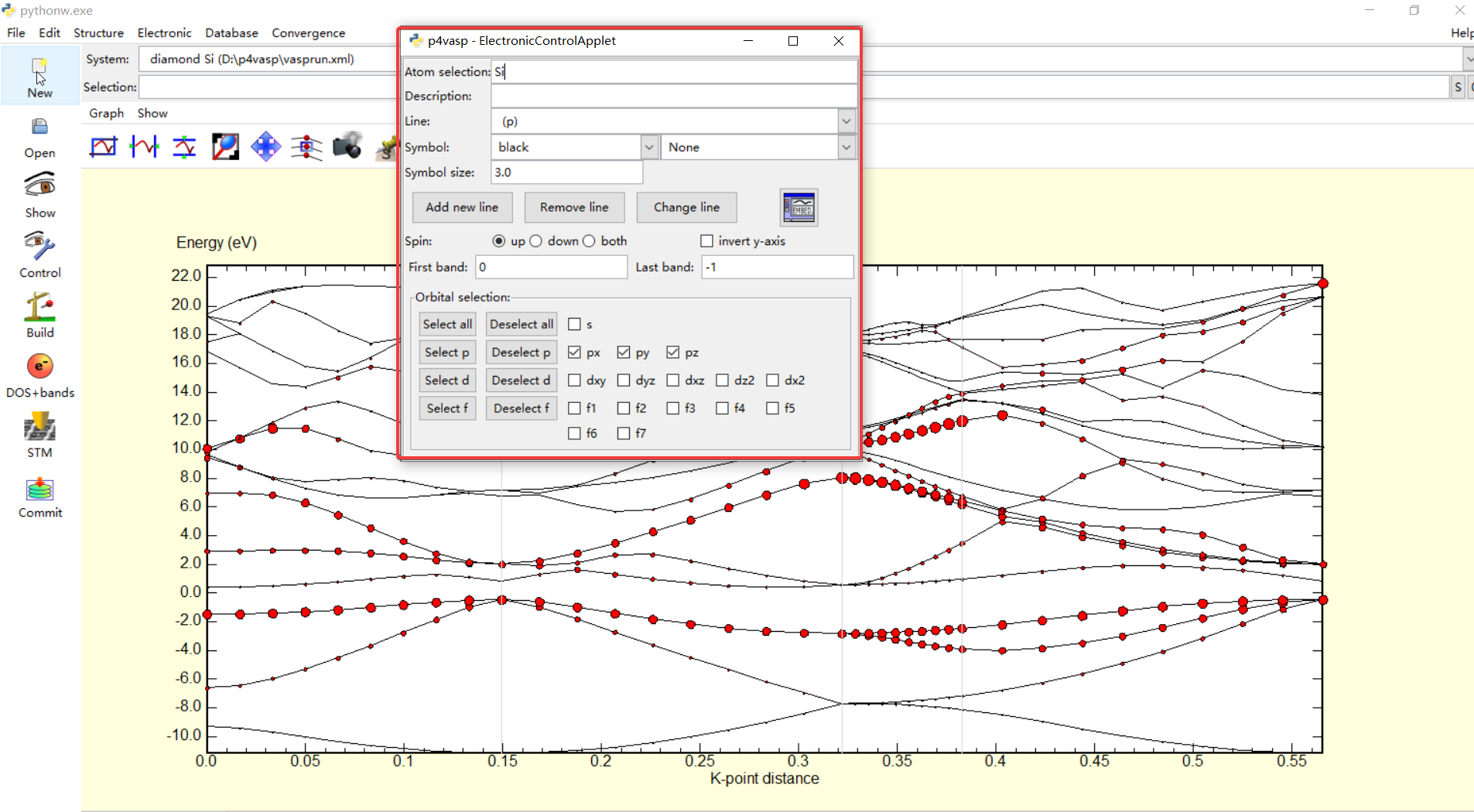
在上面,我想将Si原子的p轨道($p_x,p_y,p_z$)在能带中单独显示出来,也就是上图中红框选选中区域中的参数设置,执行Add new Line之后就发现图中的能带会被红色的圆圈单独标记出来,也就是Si的p轨道所产生的能带.同样的也可以再增加其它的一些轨道的能带,只要以颜色区分就好,在下面再把Si的s轨道产生的能带利用另外一种颜色标记出来
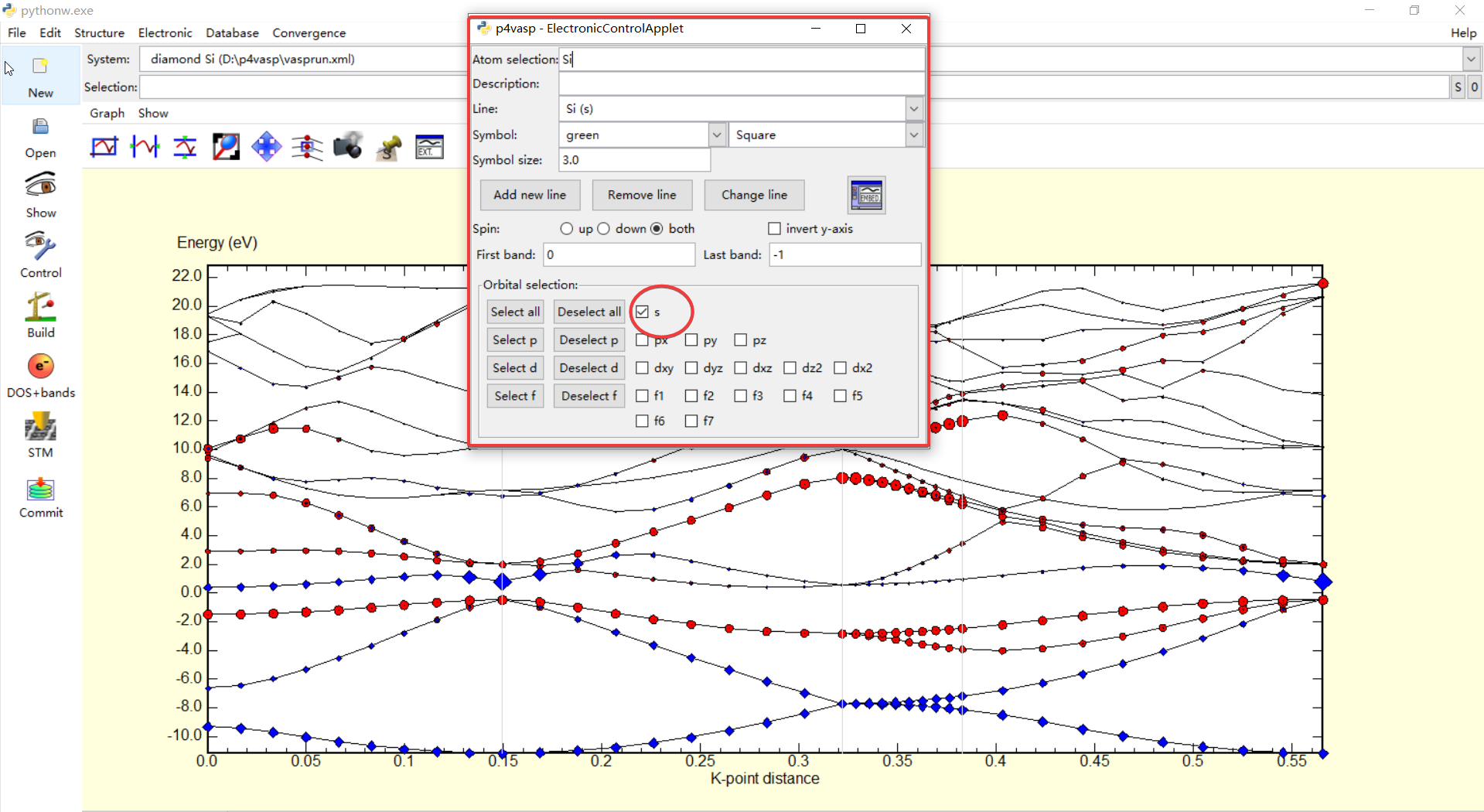
利用这种方式,就可以轻松的将原子不同轨道所产生的能带区别出来,这也正好回答了我以前的一些困惑,利用这个软件也将自己在固体物理中学习紧束缚近似时候,各个轨道在形成能带时候的一些重叠以及交叉的结构清晰的展现出来,算是对知识的进一步认识.
参考
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。
{:.info}

|
yxliphy@gmail.com |
 wechat
wechat alipay
alipay