这篇Blog整理一下自己学习使用irvsp并结合拓扑量子化学计算元素Bi的拓扑性质时候的一些笔记,其实主要就是整理计算过程。
irvsp
关于irvsp的安装和使用可以参考Irvsp学习笔记这篇博客,这里就不再重复提及了。不过唯一 需要多说的是,在这里不止要使用src_irvsp_v2_release,还需要用到src_trace_v1,所以可以在安装的时候,将这个文件夹下面的也一起安装。安装完成之后 可以在文件夹下面看到vasp2trace这个可执行文件,如下图所示
将这个可执行文件增加到你的路径中就可以
export PATH=yourpath/src_trace_v1/:$PATH
到这里准备工作就完成了,下面开始vasp的计算。
Bi第一性计算
这里就不计算晶格弛豫了,直接进行自洽计算
自洽计算
首先是控制文件INCAR
SYSTEM = Bi
ISTART = 0
ICHARG = 2
IALGO = 38
EDIFF = 1E-7
#EDIFFG = 1E-6
LREAL = False
ENCUT = 500
#NBANDS= 50
ISMEAR = 0 # -5---> metal 0,1--->semiconductor 0 for all
SIGMA = 0.05
#spin orbit coupling
LSORBIT =.TRUE.
SAXIS = 0 0 1
#ISPIN = 2
MAGMOM = 100*0.0
LORBIT = 11
LWAVE = .FALSE.
LMAXMIX = 4
#NPAR = 4
- POSCAR
generated by phonopy
1.0
2.2665000000000002 1.3085643851182867 3.9323333333333332
-2.2665000000000002 1.3085643851182867 3.9323333333333332
0.0000000000000000 -2.6171287702365733 3.9323333333333332
Bi
2
Direct
0.7659300000000000 0.7659300000000000 0.7659300000000000
0.2340700000000000 0.2340700000000000 0.2340700000000000
- KPOINTS
pymatgen 4.7.6+ generated KPOINTS with grid density = 406 / atom
0
Gamma
10 10 10
完成自洽计算之后就可以得到CHGCAR文件,利用这个文件进行下面的计算。
$\mathcal{Z}_4$
\(\kappa=\frac{1}{4}\sum_{\mathbf{k}\in\text{TRIM}}(n_\mathbf{k}^+-n_\mathbf{k}^-)\quad\text{mod}\quad 4\)
这里的$n_\mathbf{k}^+$表示在$\mathbf{k}$这个动量处,对应的反演操作本征值为$+$的能带的数量。
出了强拓扑指标,还有弱拓扑指标,其实也就是在固定某一个$k_i=\pi$,计算剩下维度在时间反演不变动量点上的宇称值
\[w_i=\frac{1}{4}\sum_{\mathbf{k}\in\text{TRIM}}^\prime(n_\mathbf{k}^+-n_\mathbf{k}^-)\quad\text{mod}\quad 2\]这里需要计算在高对称点上的反演操作算符的本征值,就需要使用到irvsp中src_irvsp_v2_release这个这个文件中的可执行文件irvsp
这里还需要使用phonopy来确定晶体结构所对应的空间群
pononopy --symmetry -c POSCAR
得到了空间群之后,就可以使用irvsp来计算所有操作在给定动量点上的本征值
irvsp
这里要想在高对称点上计算点群操作对应的本征值,还需要先将这些高对称点位置上的波函数计算出来,下面就来计算这些位置上的波函数。
- INCAR ```shell SYSTEM = Bi
ISTART = 0 ICHARG = 11
IALGO = 38 EDIFF = 1E-7 #EDIFFG = 1E-6 LREAL = False ENCUT = 500 #NBANDS= 50
ISMEAR = 0 # -5—> metal 0,1—>semiconductor 0 for all SIGMA = 0.05
#spin orbit coupling LSORBIT =.TRUE. SAXIS = 0 0 1 #ISPIN = 2 MAGMOM = 100*0.0
LORBIT = 11 LMAXMIX = 4 NPAR = 4
- POSCAR
```shell
generated by phonopy
1.0
2.2665000000000002 1.3085643851182867 3.9323333333333332
-2.2665000000000002 1.3085643851182867 3.9323333333333332
0.0000000000000000 -2.6171287702365733 3.9323333333333332
Bi
2
Direct
0.7659300000000000 0.7659300000000000 0.7659300000000000
0.2340700000000000 0.2340700000000000 0.2340700000000000
- KPOINTS
k-points 4 rec 0.00000000 0.00000000 0.00000000 1.0 0.50000000 0.50000000 0.50000000 1.0 0.50000000 0.50000000 0.00000000 1.0 0.00000000 0.50000000 0.00000000 1.0和前面自洽计算唯一不同的就是,这里要使用自洽计算得到的
CHGCAR文件,然后在KPOINTS中给出高对称点的位置,不过这个高对称点的信息在irvsp中也已经给了,如下图所示
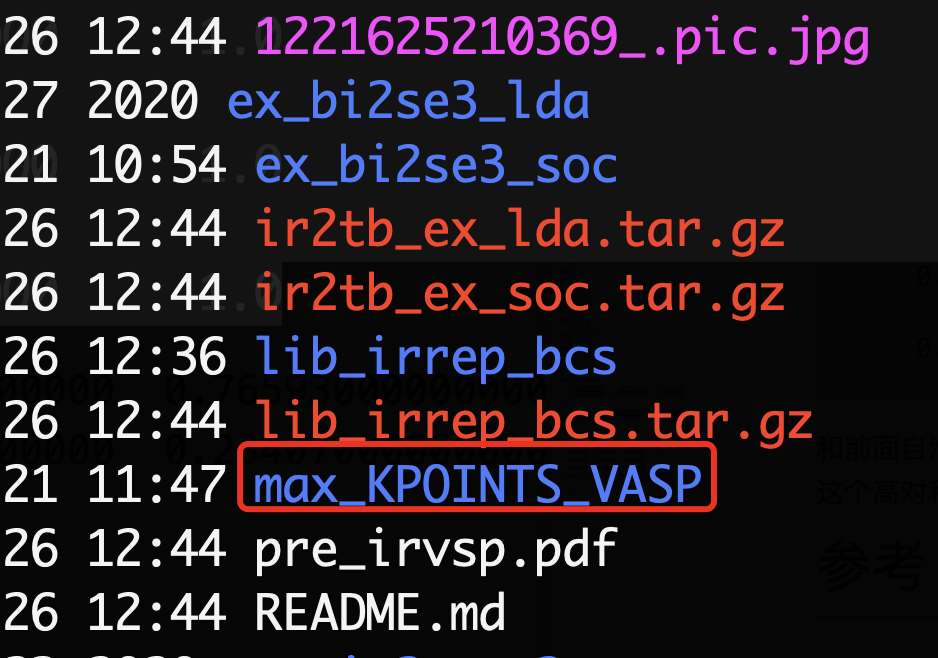
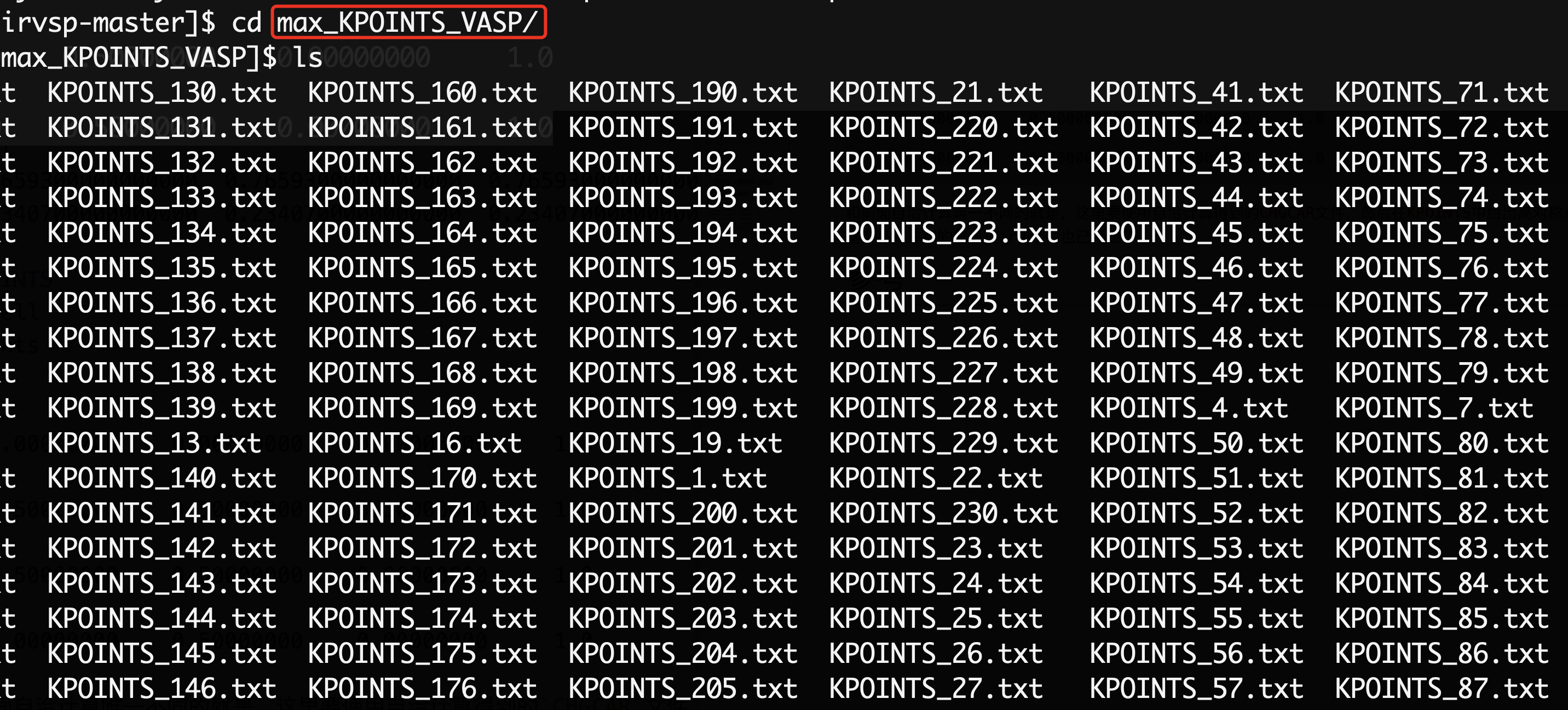
在max_KPOINTS_VASP这个文件夹中给出了每个空间群对应的高对称点的位置,比如$Bi$是166号空间群,那么找到KPOINTS_166.tx替换KPOINTS即可。做好了准备工作之后,就可以使用VASP计算这些高对称点上的波函数了,波函数的结果会保存在
WAVECAR这个文件中。
得到了波函数的的信息之后,下面就可以使用irvsp来计算高对称点上操作的本征值
irvsp -sg 166 >outir &
如果你的irvsp都配置好之后,那么执行上面的这个命令,就可以在outir中得到结果
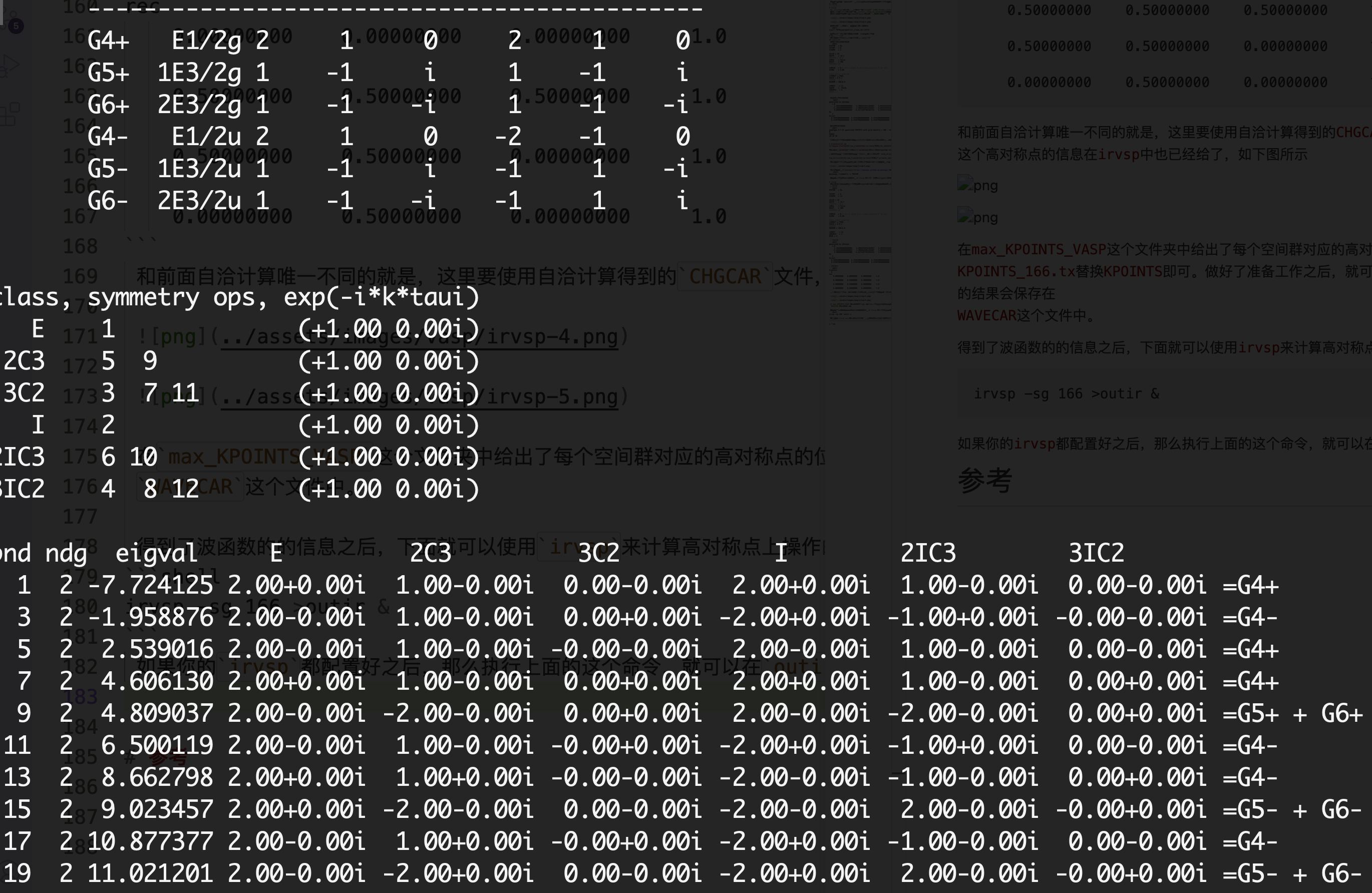
到这里也就可以使用前面$\mathcal{Z}_4$的定义来计算拓扑不变量了。
vasp2trace
其实还可以使用拓扑量子化学来进行计算,具体的理论我也还在学习中,如果有哪位懂的话,可以教教我,我在这里就只是整理一下工具的使用方法。
前面已经安装好了vasp2trace,其实在这里就非常傻瓜,在上面执行irvsp的目录中,运行
vasp2trace &
最终会得到一个trace.txt的文件

得到了这个文件之后,下面就是将这个文件上传到拓扑量子化学的一个网站上面,如下图所示
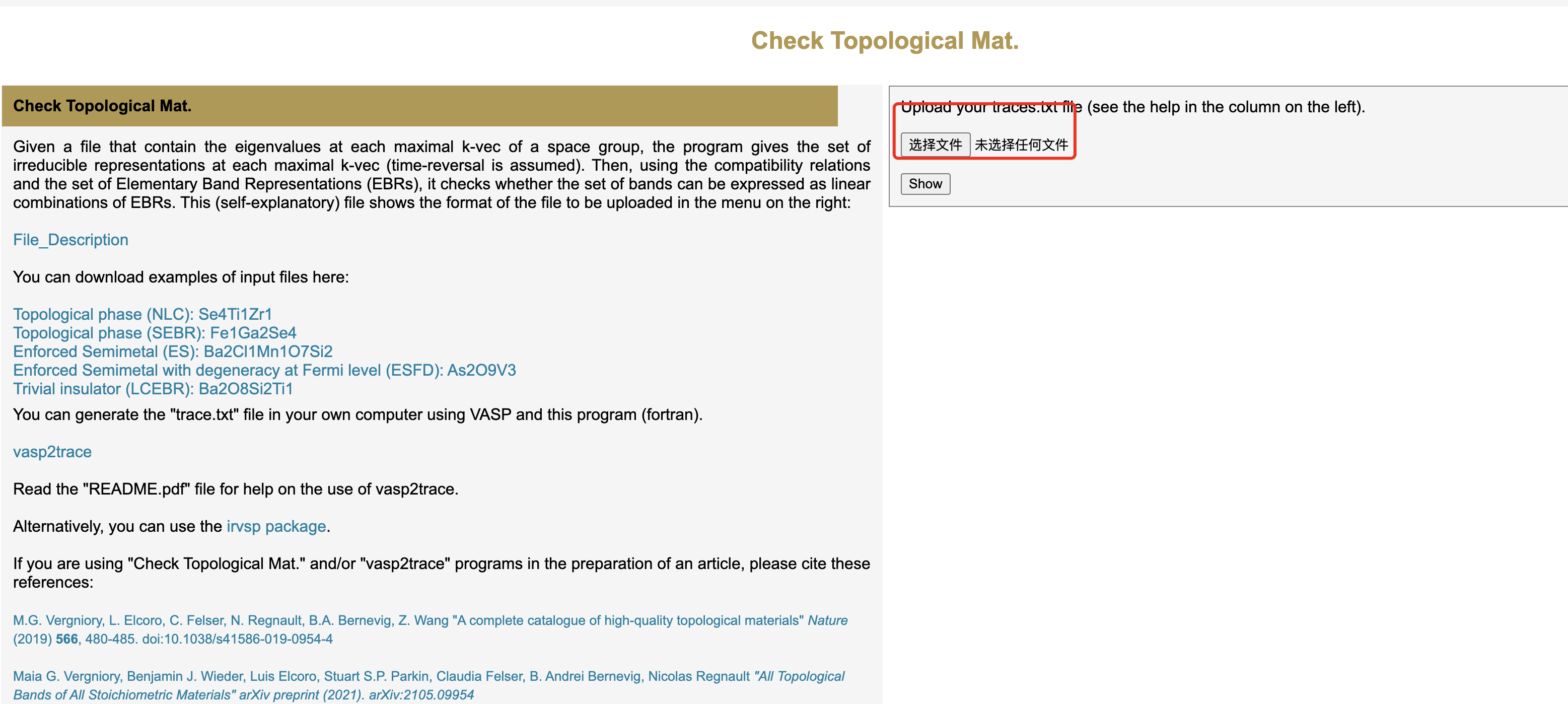
最终就可以得到下面的结果

可以看到,这里会直接计算出拓扑不变量,到这里就完事了,后面我也会具体再整理一个典型的例子来加强理论和计算的结合。
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。

|
yxli406@gmail.com |