在学习VASP的时候,看了很多教程,但看完后对一些东西的理解忘的也快,所以索性自己也通过博客的形式来把一些基本的参数输入记录一下,并加入自己对这些参数的认识,这样学习完之后还可以有笔记可查,如果忘记了还可以回来看看自己的笔记.这里我主要想把VASP最基本的4个输入文件:INCAR,POTCAR,POSCAR,KPOINTS,的最简单的输入参数进行整理,并结合自己所学的知识,尽量对这些参数的含义写出自己的理解.
INCAR
首先要说明一点,VASP的所有输入文件名都是大写的,这应该是个习惯问题,毕竟是Fortran写的程序,而Fortran最初的习惯就是用大写形式进行编程,后来应该是形成习惯了,也就一直沿用大写的方式,所以在准备这四个最基本的文件的时候,还是保持软件的习惯比较好.
INCAR是非常重要的输入文件,它决定了你要做什么,要怎么做,其中包含了很多参数,但是不必太担心,绝大多数参数都是由默认值的,如果你不清楚某些参数的具体含义,那么就不要修改它,利用提供的默认值即可.由于这个文件包含的参数很多,一一罗列并没有什么意义,所以最后我准备以VASP wiki上,最简单的氧(O)原子为实例展示一下最基本的INCAR参数.
POTCAR
这个文件包含了计算时对每个原子所使用的赝势.通常情况下,安装好VASP后就会连带着有赝势文件.
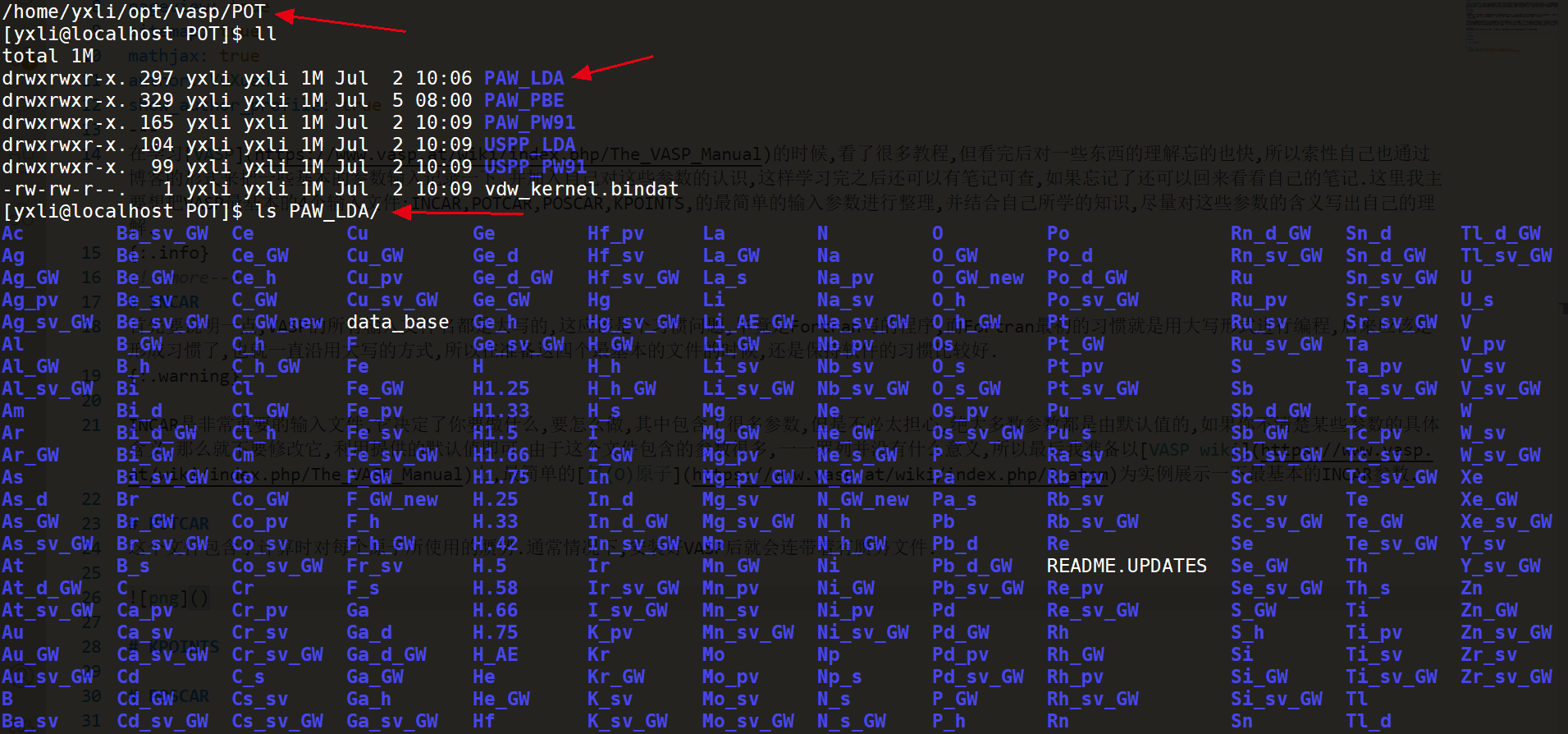
可以看到在我的vasp安装路径下有一个POT文件夹,里面包含了不同形式的赝势,而每个赝势文件夹下面则都是一些原子的赝势,赝势文件的选取就要根据自己计算的体系的性质来选取了,我也是刚开始学习,对如何选择合适的赝势文件并没有经验,就不多介绍了.通常情况你计算的体系中肯定不会只包含一种原子,那么我们这个时候就要对不同原子的赝势文件进行拼接,将它们整合到同一个文件中,并命名为POTCAR,然后将这个文件放到和INCAR相同的文件夹下面.
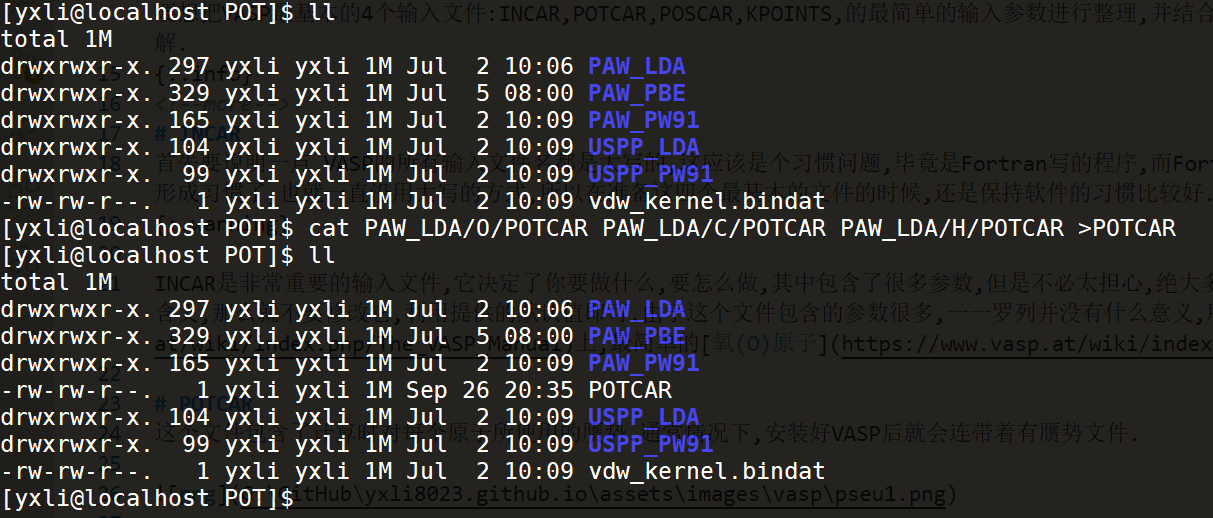
假设我的体系中含有O,C,H三种原子,那么我在这里选取的是PWA_LDA这种赝势,将这里面的三个元素的赝势POTCAR分别提取出来(cat file就是打印这个文件的所有内容),最后通过重定向符号(>)将这些本来应该输出在终端的内容重定向输入到POTCAR这个文件中,最后在当前目录下就可以看到一个POTCAR的文件.这里强调一下,在收集不同原子的赝势时,元素的顺序你是需要记住的,因为后面再POSCAR文件中设置原子个数及原子在元胞中的位置的时候,设置的顺序一定要和你构建这个赝势文件时原子的顺序相同,否则就可能导致C用的是O的赝势这种错误的情况,这时候的计算肯定不会正确.
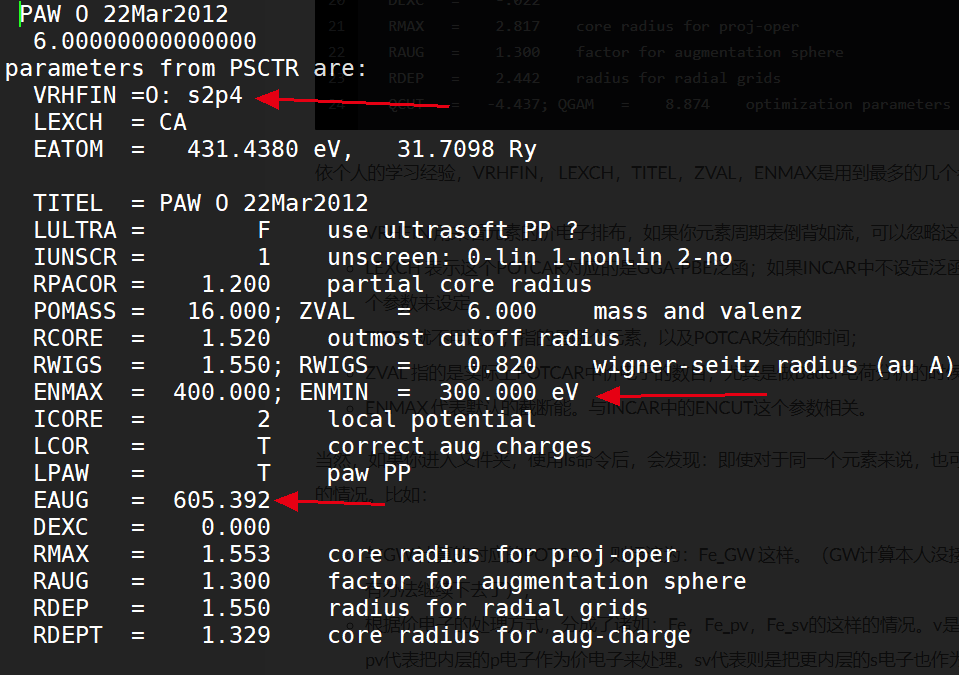
赝势文件的内容很多,这里就展示一下O原子的赝势文件中前面几行的内容,这里面具体的参数含义我也正在学习中
通常你也可以将这个VASP自带的赝势文件库放在你自己想放的位置,主要是方便你的构建赝势文件的时候方便使用它即可,同时网上也是有程序可以直接来构建赝势文件的,这个如果有机会我也会进行整理.
赝势的选取因为跟计算体系有关,所以这里就给一下官网的链接,里面都是一些在计算时对赝势的选择建议.
KPOINTS
在这里我主要是通过vasp手册上对k点的操作来学习,下面是手册上提供的k点设置示例,虽然这不是最简单的KPOINTS文件设置,但我还是想对这种方式的k点设置进行学习
Example file # 第一行就是起个名
4 # 这一行指定k点的数目
Cartesian # 这一行用来说明k点的坐标是在实空间(C)还是在倒空间(K)中给出;识别的时候只关系第一个字母,而且不区分大小写
0.0 0.0 0.0 1. # 接下来的4行就明确给出了在确定的坐标空间中4个k点的位置,这里注意到每一行都有4个数,前三个代表坐标,第四个数代表这个k点的权重
0.0 0.0 0.5 1. # 这些点的权重加起来不必一定是1,vasp会自动对这个权重进行重整化,将其调整都就会使得求和权重为1
0.0 0.5 0.5 2.
0.5 0.5 0.5 4.
Tetrahedra # 使用四面体积分方法进行进行占据计算
1 0.183333333333333 # 第一个参数描述使用四面体的数目,第二个参数描述四面体与你现在计算体系的布里渊区(BZ)体积的比值(这个比值需要自己算出来)
6 1 2 3 4 # 第一个参数描述对称简并的权重,接下来的四个参数分别对应着四面体的4个角点
在以倒空间元胞基矢来表示k点的时候
\[\vec{k}=x_1\vec{b}_1+x_2\vec{b}_2+x_3\vec{b}_3\]需要提供的就仅仅是$x_i$.如果是以直角坐标来描述k点
\[\vec{k}=\frac{2\pi}{a}(x_1,x_2,x_3)\]此时同样需要提供的就是$x_i$.这里说明一下,在直角坐标系内,基矢肯定就是直角坐标$(1,0,0),(0,1,0),(0,0,1)$,但是在倒空间中的时候,基矢则是元胞基矢,所以它们之间也是有一定的联系的.
能带计算k点选择
通常在计算体系的能带时,总是沿着高对称线进行,这里就整理一下这种高对称线上能带的计算中k点是如何选取的.
k-points along high symmetry lines # 随便起个名,最好是自己明白在干什么
40 # 40 intersections
Line-mode # 以L开头的字符,明确要计算一条线上的能带
cart # k点以直角坐标来表示(C or c开头),同样的也可以使用倒空间坐标
0 0 0 ! gamma
0 0 1 ! X
0 0 1 ! X
0.5 0 1 ! W
0.5 0 1 ! W
0 0 1 ! gamma
自动生成k点计算
Automatic mesh # 自己起名字
0 # number of k-points = 0 ->automatic generation scheme(0即代表默认自动生成k点进行计算)
Auto # fully automatic 如果字符以'a'开头,那么就说明要自动生成k格点,以Gamma(0,0,0)为中心形成Monkhorst-Pack格点.
10 ! length (l) 这个参数用来决定每个方向上取的点的数目,也就是下面这个表达式
这里的$\vec{b}_i$是元胞基矢.在确定了长度L之后,也就是上面参数中最后一行的10,那么k点网格的坐标为
\[\vec{k}=\vec{b}_{1} \frac{n_{1}}{N_{1}}+\vec{b}_{2} \frac{n_{2}}{N_{2}}+\vec{b}_{3} \frac{n_{3}}{N_{3}}, \quad n_{1}=0 \ldots, N_{1}-1 \quad n_{2}=0 \ldots, N_{2}-1 \quad n_{3}=0 \ldots, N_{3}-1\]根据固体物理的知识就知道,元胞肯定是具有一定的对称性的,所以上面的许多k点之间都是相互等价的,那么在计算的时候就可以通过对称性将很多k点进行约化,也就是不用去计算,而是简单的通过对称性就可以得到它的计算结果.
在这里同样可以人为的设置每个方向上所取格点的数量
Automatic mesh
0 # number of k-points = 0 ->automatic generation scheme
Gamma # generate a Gamma centered grid 以Gamma点为中心来生成格点
4 4 4 # subdivisions N_1, N_2 and N_3 along recipr. l. vectors 这三个参数就是来决定每个方向上的k点取值数目
0. 0. 0. # optional shift of the mesh (s_1, s_2, s_3) 这个参数用来设置在生成格点时,对位置进行一个平移
Monkhorst-Pack格点生成
在上面的参数设置中,如果把Gamma换成’M’或者’m’开头的字符,那么就采用Monkhorst-Pack方案来产生k点
\[\vec{k}=\vec{b}_{1} \frac{n_{1}+1 / 2}{N_{1}}+\vec{b}_{2} \frac{n_{2}+1 / 2}{N_{2}}+\vec{b}_{3} \frac{n_{3}+1 / 2}{N_{3}}\]在利用这个方案时,有一个条件$mod(N_i,2) = 0$,而且中心点也是偏移Gamma点的.
最后一行是一个可选参数,是对产生格点的一个平移,在Gamma与M的不同格点产生方式下,也是不同的
\[\vec{k}=\vec{b}_{1} \frac{n_{1}+s_{1}}{N_{1}}+\vec{b}_{2} \frac{n_{2}+s_{2}}{N_{2}}+\vec{b}_{3} \frac{n_{3}+s_{3}}{N_{3}}\qquad Gamma\\ \vec{k}=\vec{b}_{1} \frac{n_{1}+s_{1}+1 / 2}{N_{1}}+\vec{b}_{2} \frac{n_{2}+s_{2}+1 / 2}{N_{2}}+\vec{b}_{3} \frac{n_{3}+s_{3}+1 / 2}{N_{3}}\qquad Monkhorst-Pack\]从这个k点产生公式也可以清晰看到,对于Monkhorst-Pack方式的格点生成,相当于是以Gamma点为中心的k点产生方式平移了(0.5,0.5,0.5).
下面利用Mathematica来对这两种格点生成方式进行模拟
Gp[{N1_, N2_, N3_}, {s1_, s2_, s3_}] := Table[{(n1 + s1)/N1, (n2 + s2)/N2, (n3 + s3)/N3}, {n1, 0,
N1 - 1}, {n2, 0, N2 - 1}, {n3, 0, N3 - 1}]
Mp[{N1_, N2_, N3_}, {s1_, s2_, s3_}] := Table[{(n1 + s1 + 1/2)/N1, (n2 + s2 + 1/2)/N2, (n3 + s3 + 1/2)/
N3}, {n1, 0, N1 - 1}, {n2, 0, N2 - 1}, {n3, 0, N3 - 1}]
p1 = Flatten[Gp[{3, 3, 3}, {0, 0, 0}], 2];
p2 = Flatten[Mp[{3, 3, 3}, {0, 0, 0}], 2];
g1 = Graphics3D[{PointSize[0.05], Red, Point[p1]}, Axes -> True, AxesStyle -> Directive[Black, 20, Bold]];
g2 = Graphics3D[{PointSize[0.05], Blue, Point[p2]}];
Show[g1, g2, ImageSize -> Large]
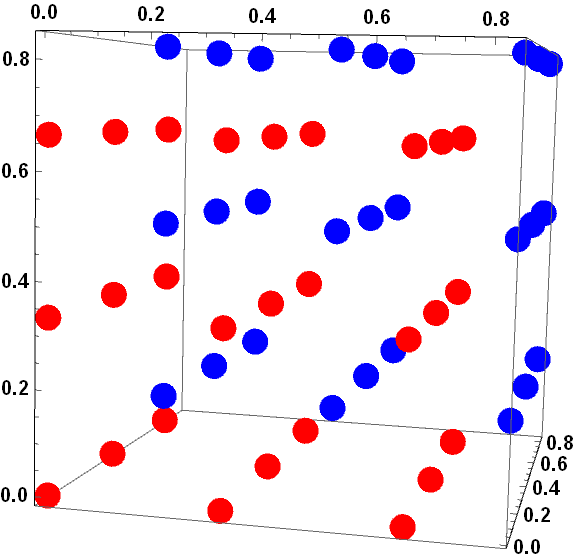
蓝色和红色就分别代表了以Gamma点为中心和Monkhorst-Pack方式生成k点的位置.
POSCAR
POSCAR文件包含了晶格的几何结构和离子的位置,同时里面还可以选择离子的初始速度以及相关的预测修正,并用来做分子动力学模拟.这个文件在前面的一篇博客里面已经详细的讨论过了,VASP输入文件POSCAR简单设置以及两种坐标间联系
Cubic BN # 这里就是给你要计算的体系起个名字
3.57 # 这是一个缩放系数,会对所有的格矢和原子坐标进行缩放
0.0 0.5 0.5 # 这三行就是组成格子的三条边的矢量坐标信息,缩放系数就在这里起作用
0.5 0.0 0.5 # 利用缩放系数可以得到,格子第二条边的矢量为(0.5*3.57,0,0.5*3.57)
0.5 0.5 0.0 # 这三个矢量就是用来确定元胞的基矢
1 1 # 计算的系统中每个原子的数量,譬如FeSe就表示体系统有一个Fe原子,一个Se原子
Selective dynamics # 这个选项用来设置在离子弛豫过程中原子的位置是否要保持不动(这个一个可选参数,默认下也可以不设置)
Cartesian # 接下来的三行是关于分子动力学的设置,我不懂
0.00 0.00 0.00 T T F
0.25 0.25 0.25 F F F
Cartesian # 这里就是来设置坐标是直角坐标,通常都只是识别第一个字母,但是为了明确一般就写成Cartesian这个易懂的单词
0.01 0.01 0.01 # 第一个原子在元胞中的位置
0.00 0.00 0.00 # 第二个原子在元胞中的位置
# 在设置原子的坐标位置的时候,一定要和前面原子的数目一致.
在设置体系中原子数量的时候,这个一定要和你在构建赝势文件时候的元素顺序一致,这个问题在前面赝势文件构建的时候也是提及到的.
VESTA生成POSCAR
VESTA可以用来对晶体结构进行可视化,如果已经有了现成的晶体结构,那么可以通过软件来直接生成原子的直角坐标或者分数坐标.下面我通过最简单的NaCl晶体结构展示一下如何生成它的分数坐标和直角坐标.
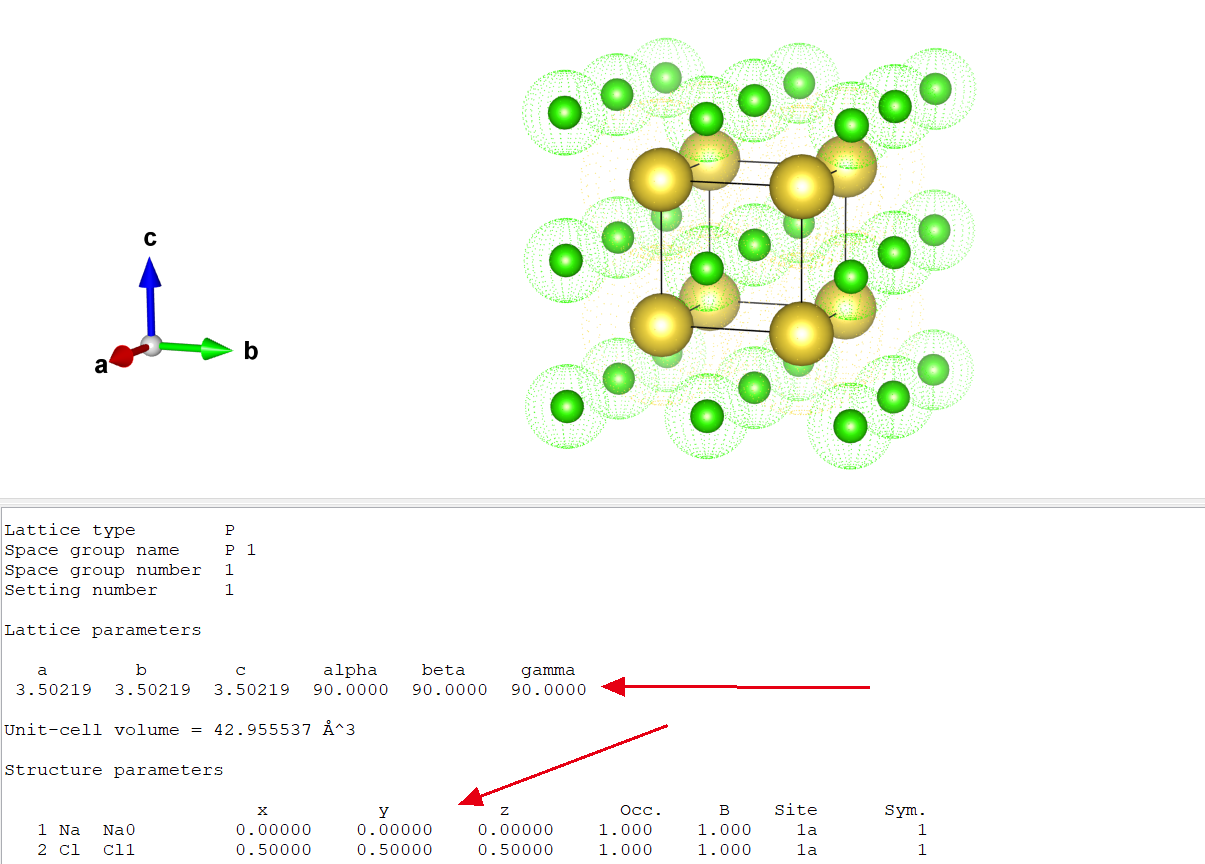
将格式为cif的晶体结构文件打开之后,上半部分就是晶体中原子的排列位置,下面就是具体的参数信息,包括不同方向上的元胞基矢的大小,以及一些其他的参数.
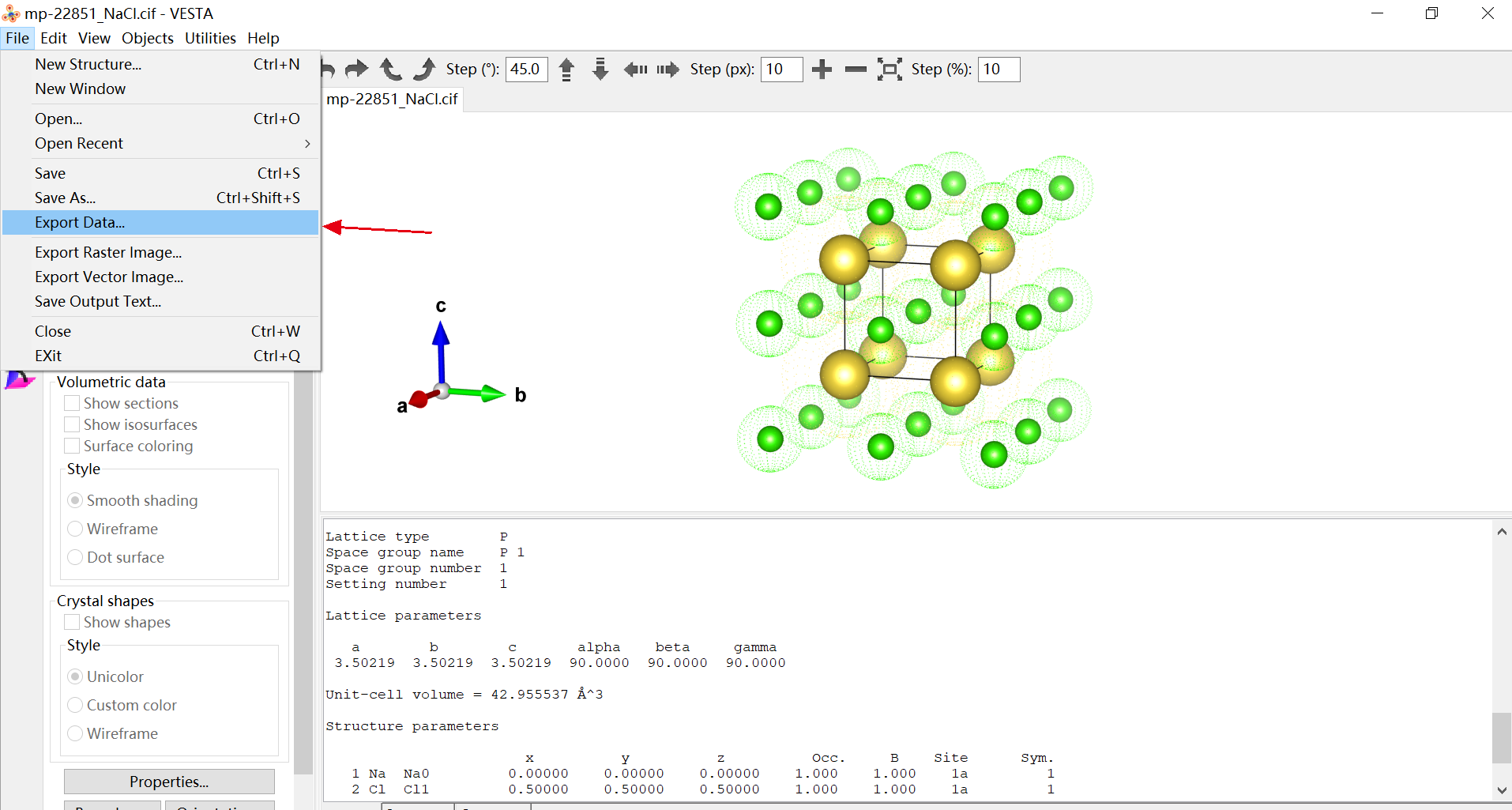
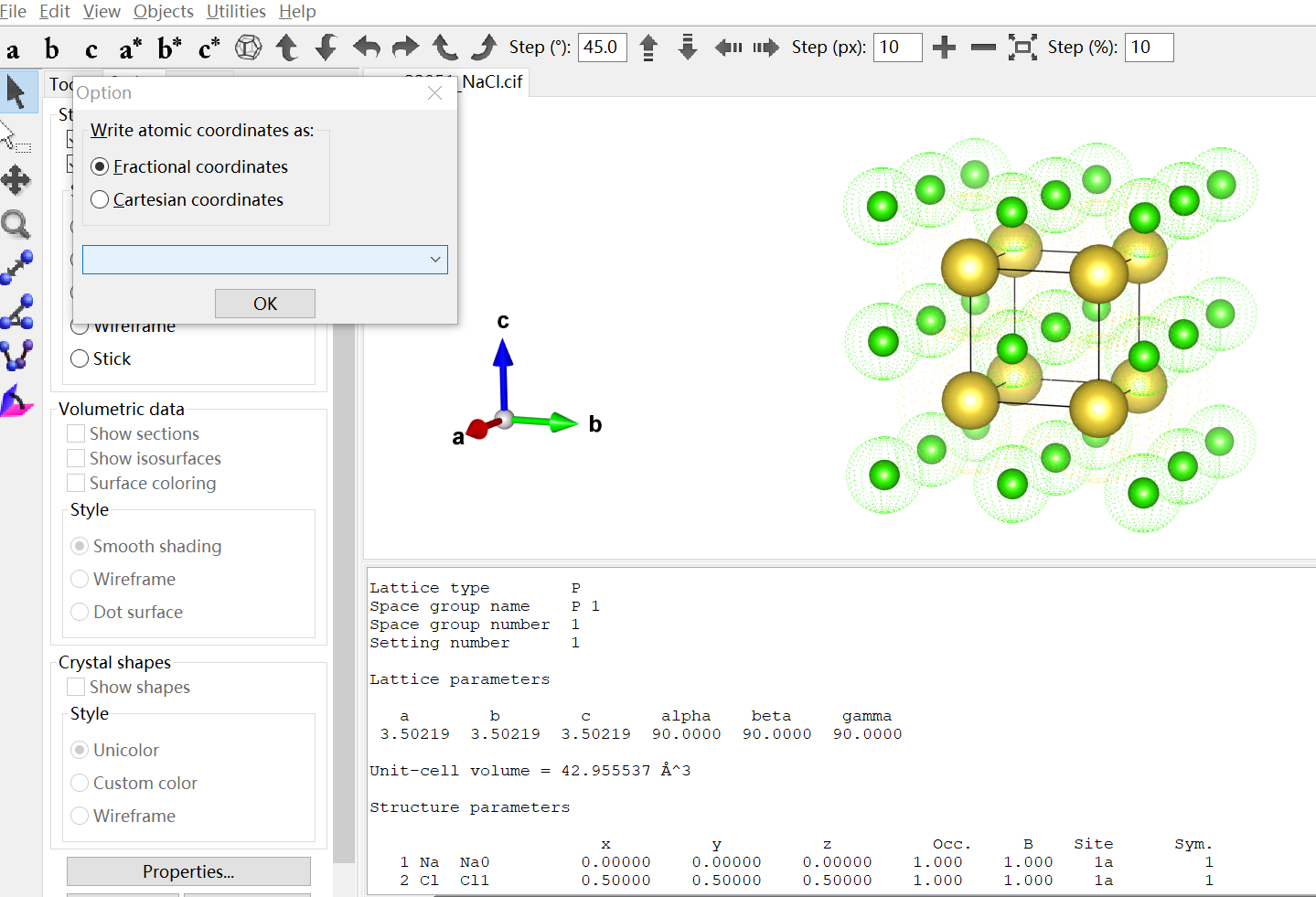
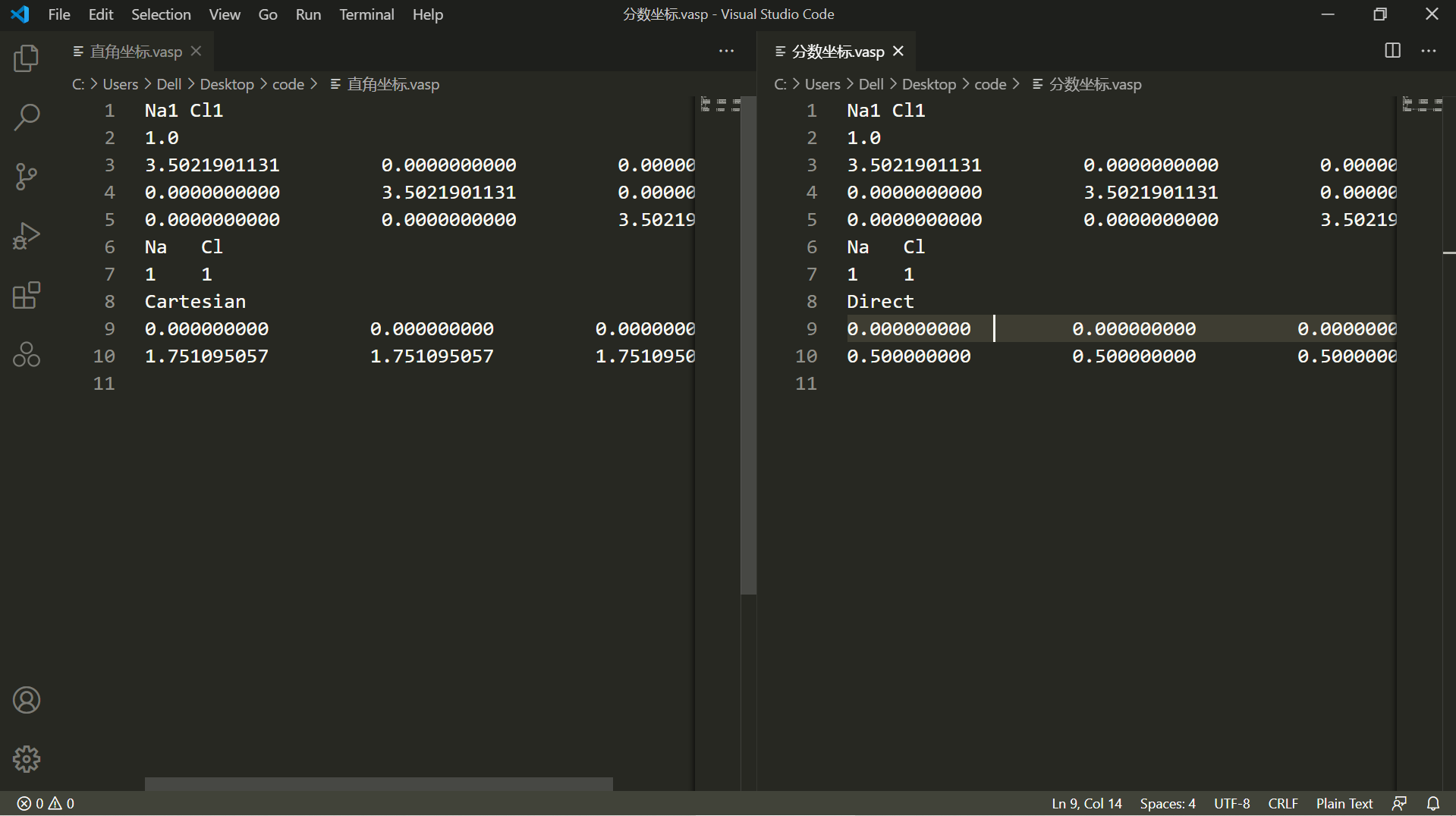
如上图操作所示,在导出数据进行保存的时候,就可以选则是哪种方式进行保存.导出后两种坐标下的内容如下图所示
氧(O)原子实例
POSCAR
O atom in a box
1.0 ! universal scaling parameters(缩放系数,认为是晶格常数即可)
8.0 0.0 0.0 ! lattice vector a(1) 元胞的三个基矢,此时一个元胞中只有一个O原子
0.0 8.0 0.0 ! lattice vector a(2)
0.0 0.0 8.0 ! lattice vector a(3)
1 ! number of atoms
cart ! positions in cartesian coordinates
0 0 0 ! 一个氧原子在坐标原点
INCAR
SYSTEM = O atom in a box !给你计算的系统起一个名字
ISMEAR = 0 ! Gaussian smearing,这个参数不同的值对应着不同的方法,对于分子和原子体系,设置为0即可.再详细的解释可以参考手册
KPOINTS
Gamma-point only
0
Monkhorst Pack
1 1 1
0 0 0
至于赝势文件的构建和前面讲的就是完全相同的,只要通过cat将赝势库中自己想要的O原子的赝势文件复制到和这三个文件相同的文件夹目录下即可.
运行vasp
我是在自己组里的小服务器上自己安装的vasp,对于如何在超算等大型服务器上如何运行vasp还不是很懂,所以我就在这里提供一下我自己在组里小服务器上运行vasp的命令
nohup mpirun -np 10 vasp
nohup是linux自己的命令,是为了让程序在后台运行,会有一个默认输出文件nohup.out,而mpirun -np 10表示我想利用10个核来实现该计算.因为O原子这个体系非常简单,而且在INCAR中设置的参数也很少,所以计算很快就可以执行完毕,得到一大堆的输出文件,这些输出的内容我也正在学习中.
参考
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。

|
yxli406@gmail.com |