VASP计算Graphene能带
最近看了很多和第一性原理计算相关的资料,知识总是要和实践结合,这里就利用VASP来计算一下Graphene的能带,作为研究很透彻的一个体系,起码在$\Gamma$点的Dirac锥的结构势清楚了,所以就拿这个体系来入门.下面的内容都是我最为一个初学者的理解,其中应该会包含一些错误的理解,希望可以指出.
{:.info}
1.晶格弛豫
在对一个体系进行计算时,首先是要确定你的这个体系时稳定的,所以要先进行晶格弛豫,我理解的就是晃动一下整个晶格,重要保证它有一定的稳定性,不至于轻微的晃动就变成另外一种结构.这里这计算的时2维的Graphene,虽然从结构上看确实好像有一定稳定性,但是还是要进行弛豫计算.
INCAR
1 | ISTART=0 # 参数0代表开始一个全新的计算 |
POSCAR
关于POSCAR的设置可以参考这篇博客VASP基本输入文件准备1
2
3
4
5
6
7
8
9
10graphene
1.0
2.4677236 0.0000000 0.0000000
-1.2338618 2.1371113 0.0000000
0.0000000 0.0000000 15.0000000
C
2
Direct
0.00000000 0.00000000 0.25000000
0.66666667 0.33333333 0.25000000
KPOINTS
1 | AUTO |
晶格弛豫过程主要是为了确定晶格的一个稳定结构,计算完成之后需要的只是CONTCAR文件,它包含了最终计算得到的稳定结果
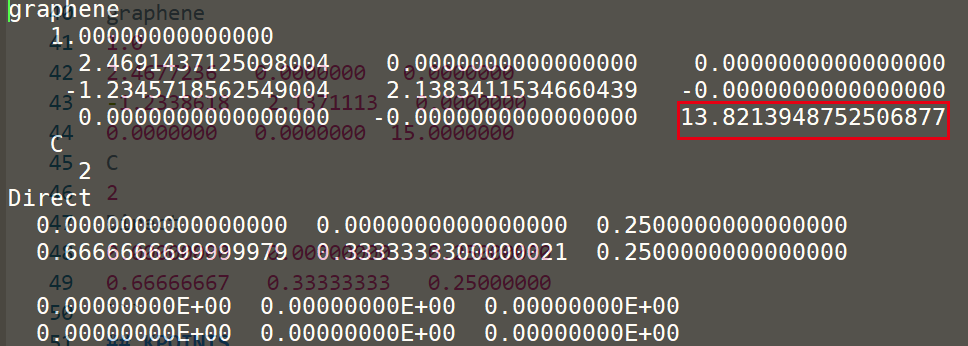
这里第三个基矢发生了一些小的变化,但是这里计算的是2维Graphene,还是将它修改维最初的15.00000
自洽计算
完成了晶格弛豫稳定性计算之后,接下来就进行自洽计算,这个时候需要将上一步计算得到的CONTCAR复制为POSCAT来进行自洽计算
INCAR
1 | ISTART = 0 # 还是要重新开始一个计算 |
KPOINTS
1 | AUTO |
自洽计算完成之后会得到电荷密度文件CHGCAR,利用这个文件来进行之后的能带计算.
能带计算
利用前两步计算得到的CHGCAR以及CONTCAR文件,将CONTCAR复制为POSCAR,进行能带计算.
INCAR
1 | ICHARG = 11 # Read from CHGCAR 从现有的文件中读取电荷密度 |
KPOINTS
1 | K-points for bandstructure G-K-M-G |
执行完毕之后即可以得到最终的结果vasprun.xml,可以通过p4vasp来查看能带结果.
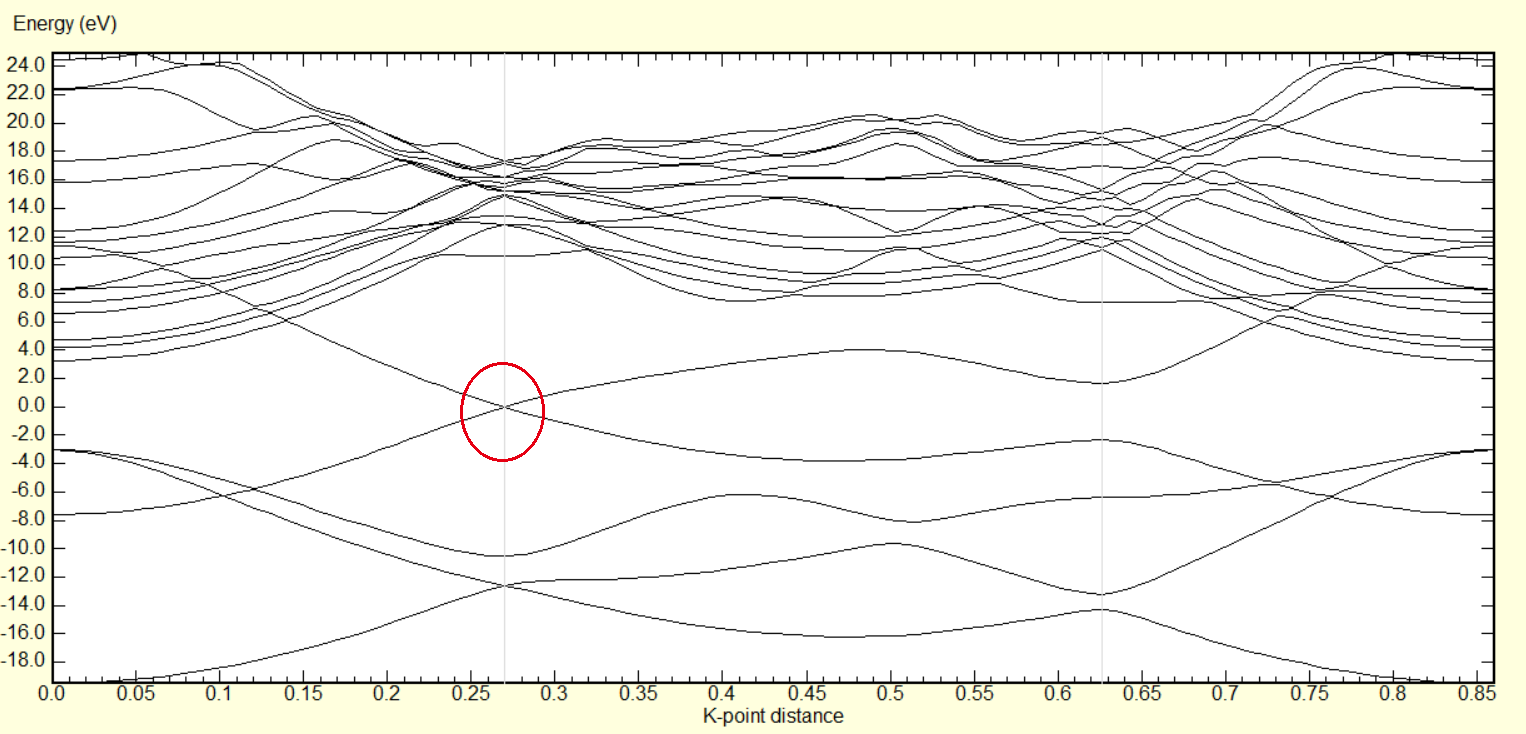
计算平台
我这里是在并行云的超算上进行了,自己提供VASP的包之后,平台会有人进行编译配置,完成之后会提供一个提交作业的脚本给你,展示一下我的1
2
3
4
5
6
7
8!/bin/bash
SBATCH -p amd_256
SBATCH -N 1
SBATCH -n 64
source /public3/soft/other/module.sh
module load mpi/intel/17.0.5-cjj-public3
export PATH=/public3/home/username/package/VASP/vasp.6.1.0/bin:$PATH
srun vasp_std
提交作业的时候,将这个脚本(比如名称为job.sh)复制到你的文件夹里面,然后执行
sbatch job.sh
关于脚本与服务器的其他内容,如果取申请自然会有相关的资料给你.
参考
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。
{:.info}

|
yxliphy@gmail.com |
 wechat
wechat alipay
alipay