$Bi_2Se_3$第一性计算结果重复
学习第一性计算也有一段时间了,这里利用VASP+Wannier90+WannierTools来完整复现一下$Bi_2Se_3$这个材料的一些拓扑性质.
{:.info}
先整理下大致的流程,首先通过VASP自洽计算,得到收敛结果. 第二步结合Wannier90得到Tight binding模型的数据信息. 第三步就是在第二步的基础上得到利用wannier90_hr.dat来计算拓扑相关的性质.
VASP
VASP的计算中需要分两步进行, 首先不考虑SOC进行一次电子自洽, 这里我是直接使用WannierTools文档中$Bi_2Se_3$的晶体结构来计算的, 所以并没有进行结构优化方面的计算.
No-SOC
VASP计算需要的文件内容如下
- INCAR
1 | SYSTEM = NaCl |
POSCAR
1
2
3
4
5
6
7
8
9
10
11
12
13Bi2Se3
1.0
-2.069 -3.583614 0.000000
2.069 -3.583614 0.000000
0.000 2.389075 9.546667
Bi Se
2 3
Direct
0.3990 0.3990 0.6970
0.6010 0.6010 0.3030
0 0 0.5
0.2060 0.2060 0.1180
0.7940 0.7940 0.8820KPOINTS
1
2
3
4
5Monkhorst Pack
0
G
12 12 3
0 0 0- POTCAR
至于赝势文件, 自行准备, 没有什么需要强调的.
准备好文件之后, 开始计算即可, 计算收敛之后会得到CHGCAR文件, 这个文件用来进行打开SOC时的收敛计算.
OPEN-SOC
新建一个文件夹, 将上一步计算得到的CHGCAR复制过来, 并将原来的INCAR进行修改
- INCAR
1 | SYSTEM = NaCl |
其余的三个输入文件都不需要改动, 然后进行自洽收敛.
Wannier90
接下来就是利用Wannier90来进行关于Tight Binding相关的计算, 还是在No-SOC自洽计算的基础上, 新建文件夹复制其CHGCAR文件, 修改INCAR如下
- INCAR
1 | SYSTEM = NaCl |
然后设置wannier90.win文件来控制投影计算
1 | num_wann = 30 # 设置需要投影的Wannier轨道 |
这里需要说明一下num_wann这个参数的设置,首先可以知道元胞内共有5个原子, 而且每个原子都需要由三个轨道投影, 所以一共有$3\times 5=15$个轨道, 但是此时需要考虑自旋, 则共有$3\times 5\times2=30$个轨道需要投影.
{:.warning}
准备好INCAR,POSCAR,POTCAR,KPOINTS,wannier90.win着四个文件之后, 就可以开始计算了. 相比于前面只需要修改INCAR文件中的内容即可. 完成计算之后可以得到wannier90.amn,wannier90.chk,wannier90.eig,wannier90.mmn,wannier90.wout,wannier90_wsvec.dat这些文件.
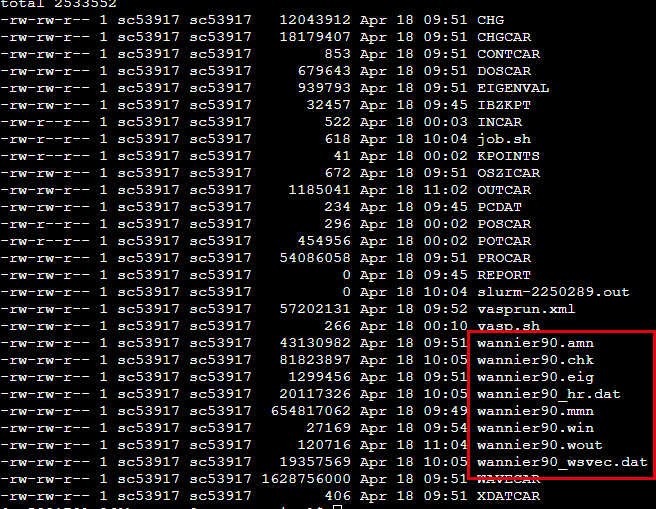
得到这些文件之后,开始利用Wannier90计算Tight Bind所需要的的数据,执行1
wannier90.x wannier90
可以最终得到wannier90_hr.dat这个文件,到此Wannier90的计算结束, 之后就可以利用这个数据来进行拓扑方面的计算.
WannierTools
下面进行拓扑方面的计算,将计算得到的wannier90_hr.dat单独复制到一个文件夹, 设置WannierTools所需要的的wt.in1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164&TB_FILE
Hrfile = 'wannier90_hr.dat'
Package = 'VASP' ! obtained from VASP, it could be 'VASP', 'QE', 'Wien2k', 'OpenMx'
/
LATTICE
Angstrom
-2.069 -3.583614 0.000000 ! crystal lattice information
2.069 -3.583614 0.000000
0.000 2.389075 9.546667
ATOM_POSITIONS
5 ! number of atoms for projectors
Direct ! Direct or Cartisen coordinate
Bi 0.3990 0.3990 0.6970
Bi 0.6010 0.6010 0.3030
Se 0.0000 0.0000 0.5000
Se 0.2060 0.2060 0.1180
Se 0.7940 0.7940 0.8820
PROJECTORS
3 3 3 3 3 ! number of projectors
Bi px py pz ! projectors
Bi px py pz
Se px py pz
Se px py pz
Se px py pz
SURFACE ! Specify surface with two vectors, see doc
1 0 0
0 1 0
!> bulk band structure calculation flag
&CONTROL
BulkBand_calc = T
BulkBand_points_calc = T
DOS_calc = T
SlabBand_calc = T
SlabBandWaveFunc_calc = T
SlabBand_plane_calc = T
WireBand_calc = T
SlabSS_calc = T
SlabArc_calc = T
SlabQPI_calc = T
Z2_3D_calc = T
SlabSpintexture_calc = T
Wanniercenter_calc = T
/
&SYSTEM
NSLAB = 4 ! for thin film system
NSLAB1= 2 ! nanowire system
NSLAB2= 2 ! nanowire system
NumOccupied = 15 ! NumOccupied
SOC = 1 ! soc
E_FERMI = 4.9687 ! e-fermi, a global shift of the energy levels
surf_onsite= 0.0 ! surf_onsite
/
&PARAMETERS
Eta_Arc = 0.001 ! infinite small value, like brodening
E_arc = 0.0 ! energy level for contour plot of spectrum
OmegaNum = 400 ! omega number
OmegaMin = -0.6 ! energy interval
OmegaMax = 0.5 ! energy interval
Nk1 = 101 ! number k points odd number would be better
Nk2 = 101 ! number k points odd number would be better
Nk3 = 101 ! number k points odd number would be better
NP = 5 ! number of principle layers
Gap_threshold = 0.01 ! threshold for FindNodes_calc output
/
KPATH_BULK ! k point path
4 ! number of k line only for bulk band
G 0.00000 0.00000 0.0000 Z 0.00000 0.00000 0.5000
Z 0.00000 0.00000 0.5000 F 0.50000 0.50000 0.0000
F 0.50000 0.50000 0.0000 G 0.00000 0.00000 0.0000
G 0.00000 0.00000 0.0000 L 0.50000 0.00000 0.0000
KPATH_SLAB
2 ! numker of k line for 2D case
K 0.33 0.67 G 0.0 0.0 ! k path for 2D case
G 0.0 0.0 M 0.5 0.5
KPLANE_SLAB
-0.1 -0.1 ! Original point for 2D k plane
0.2 0.0 ! The first vector to define 2D k plane
0.0 0.2 ! The second vector to define 2D k plane for arc plots
KPLANE_BULK
0.00 0.00 0.50 ! Original point for 3D k plane
1.00 0.00 0.00 ! The first vector to define 3d k space plane
0.00 0.50 0.00 ! The second vector to define 3d k space plane
KCUBE_BULK
-0.50 -0.50 -0.50 ! Original point for 3D k plane
1.00 0.00 0.00 ! The first vector to define 3d k space plane
0.00 1.00 0.00 ! The second vector to define 3d k space plane
0.00 0.00 1.00 ! The third vector to define 3d k cube
WANNIER_CENTRES ! copy from wannier90.wout
Cartesian
0.000001 -1.194434 6.617259
0.000041 -1.194660 6.617233
-0.000003 -1.188083 6.631973
0.000159 -1.188134 6.632086
0.000080 -1.200875 6.632263
-0.000066 -1.200732 6.632358
0.000027 -3.583694 2.929604
-0.000171 -3.583517 2.929473
0.000082 -3.590239 2.914729
0.000047 -3.589978 2.914565
-0.000105 -3.577213 2.914447
0.000111 -3.577363 2.914243
0.000003 1.194539 4.773298
0.000006 1.194535 4.773338
0.000042 1.194556 4.773340
-0.000034 1.194539 4.773342
-0.000012 1.194561 4.773333
-0.000007 1.194524 4.773332
0.000026 -1.194484 1.121831
-0.000013 -1.194453 1.121916
-0.000005 -1.212092 1.141185
0.000003 -1.212120 1.141212
0.000051 -1.177254 1.142222
0.000009 -1.177300 1.142218
-0.000003 -3.583717 8.424777
-0.000007 -3.583669 8.424788
-0.000030 -3.566052 8.405460
-0.000054 -3.566017 8.405465
-0.000042 -3.600902 8.404447
0.000034 -3.600867 8.404449
0.000001 -1.194434 6.617259
0.000041 -1.194660 6.617233
-0.000003 -1.188083 6.631973
0.000159 -1.188134 6.632086
0.000080 -1.200875 6.632263
-0.000066 -1.200732 6.632358
0.000027 -3.583694 2.929604
-0.000171 -3.583517 2.929473
0.000082 -3.590239 2.914729
0.000047 -3.589978 2.914565
-0.000105 -3.577213 2.914447
0.000111 -3.577363 2.914243
0.000003 1.194539 4.773298
0.000006 1.194535 4.773338
0.000042 1.194556 4.773340
-0.000034 1.194539 4.773342
-0.000012 1.194561 4.773333
-0.000007 1.194524 4.773332
0.000026 -1.194484 1.121831
-0.000013 -1.194453 1.121916
-0.000005 -1.212092 1.141185
0.000003 -1.212120 1.141212
0.000051 -1.177254 1.142222
0.000009 -1.177300 1.142218
-0.000003 -3.583717 8.424777
-0.000007 -3.583669 8.424788
-0.000030 -3.566052 8.405460
-0.000054 -3.566017 8.405465
-0.000042 -3.600902 8.404447
0.000034 -3.600867 8.404449
上面的设置中WannierCenter的数据可以通过前一步计算得到的wannier90.wout中得到.准备好了wannier90_hr.dat,wt.in这两个文
件之后, 就可以开始拓扑性质方面的计算了,至于如何控制计算,可以自行参考WannierTools的帮助手册, 或者查看我其他相关WannierTools的
计算博客.
这里在进行计算的时候, 需要设置费米能量, 这个值需要我们从自洽结果的OUTCAR文件中寻找1
grep E-fermi OUTCAR
得到费米能并设置好之后,执行1
wt.x wt.in
开始计算,计算结束后会有一堆.gnu的文件,这是gnuplot绘图的命令文件,这里提供一个脚本,可以批量执行1
2
3
4
5
6
7!/bin/sh
============ get the file name ===========
Folder_A=$(pwd)
for file_a in ${Folder_A}/*.gnu
do
gnuplot $file_a
done
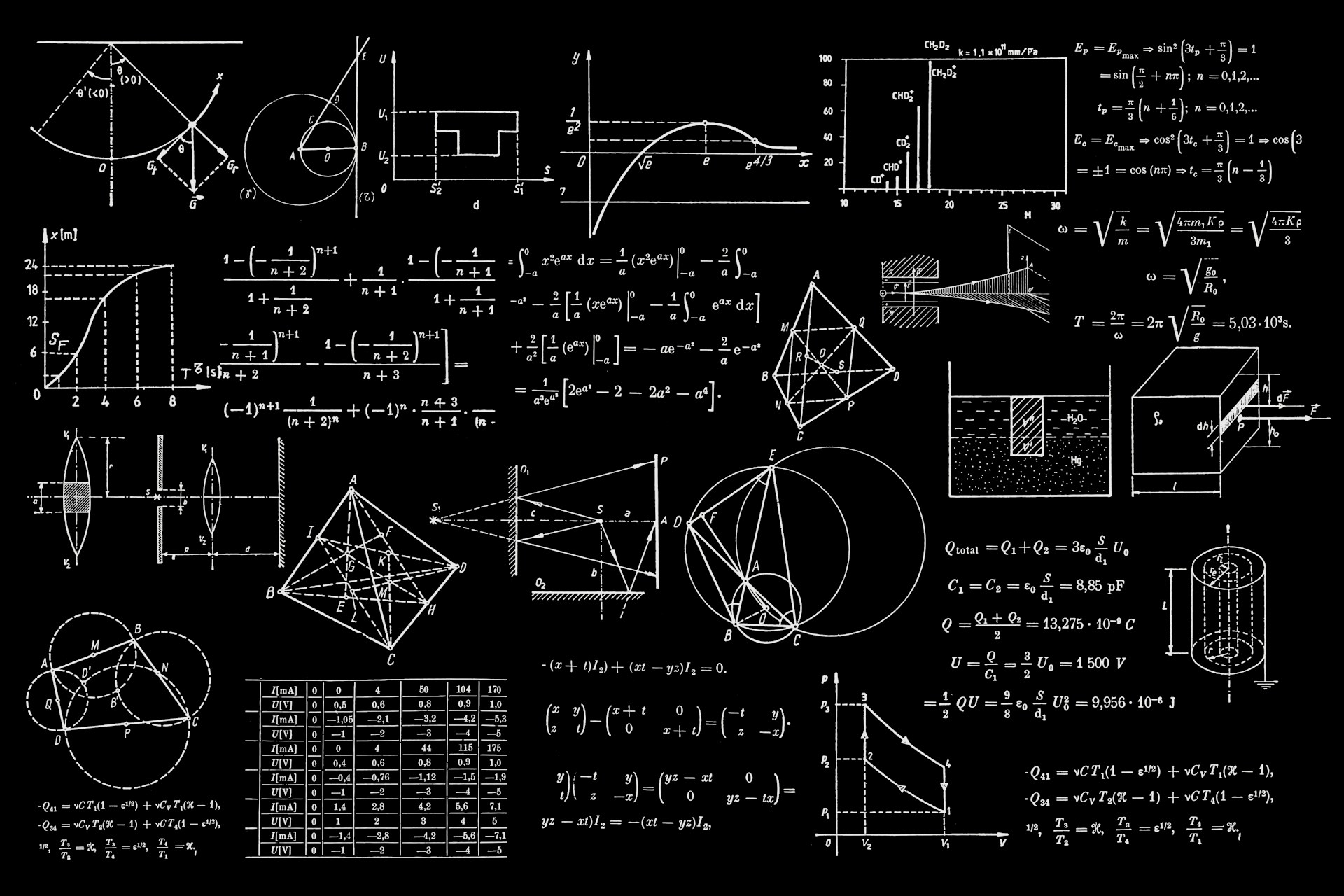
代码的本意就是对所有后缀为.gnu的文件, 执行gnuplot命令, 这样最后就可以得到所有的绘图结果了.下面是一些计算的结果
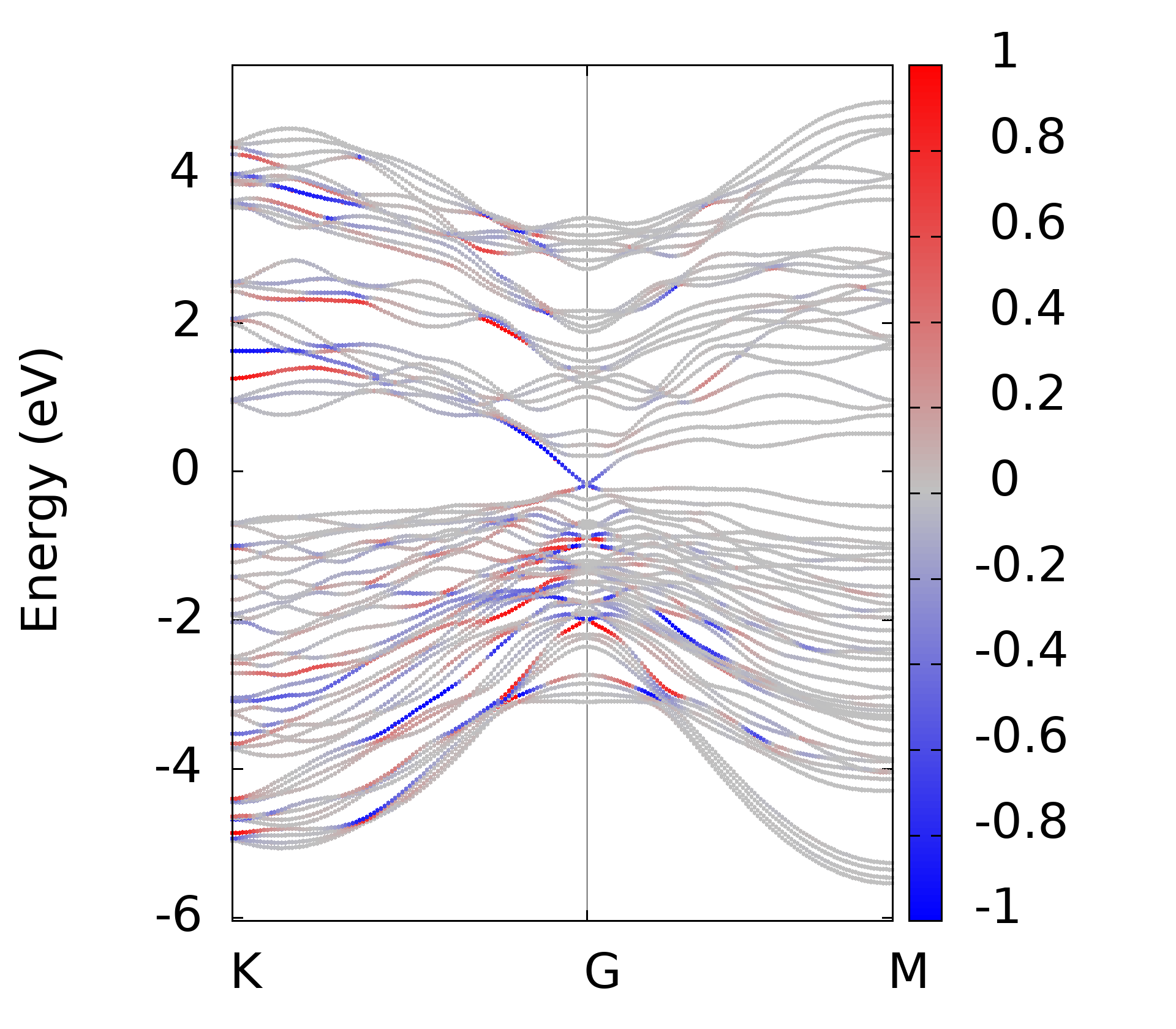
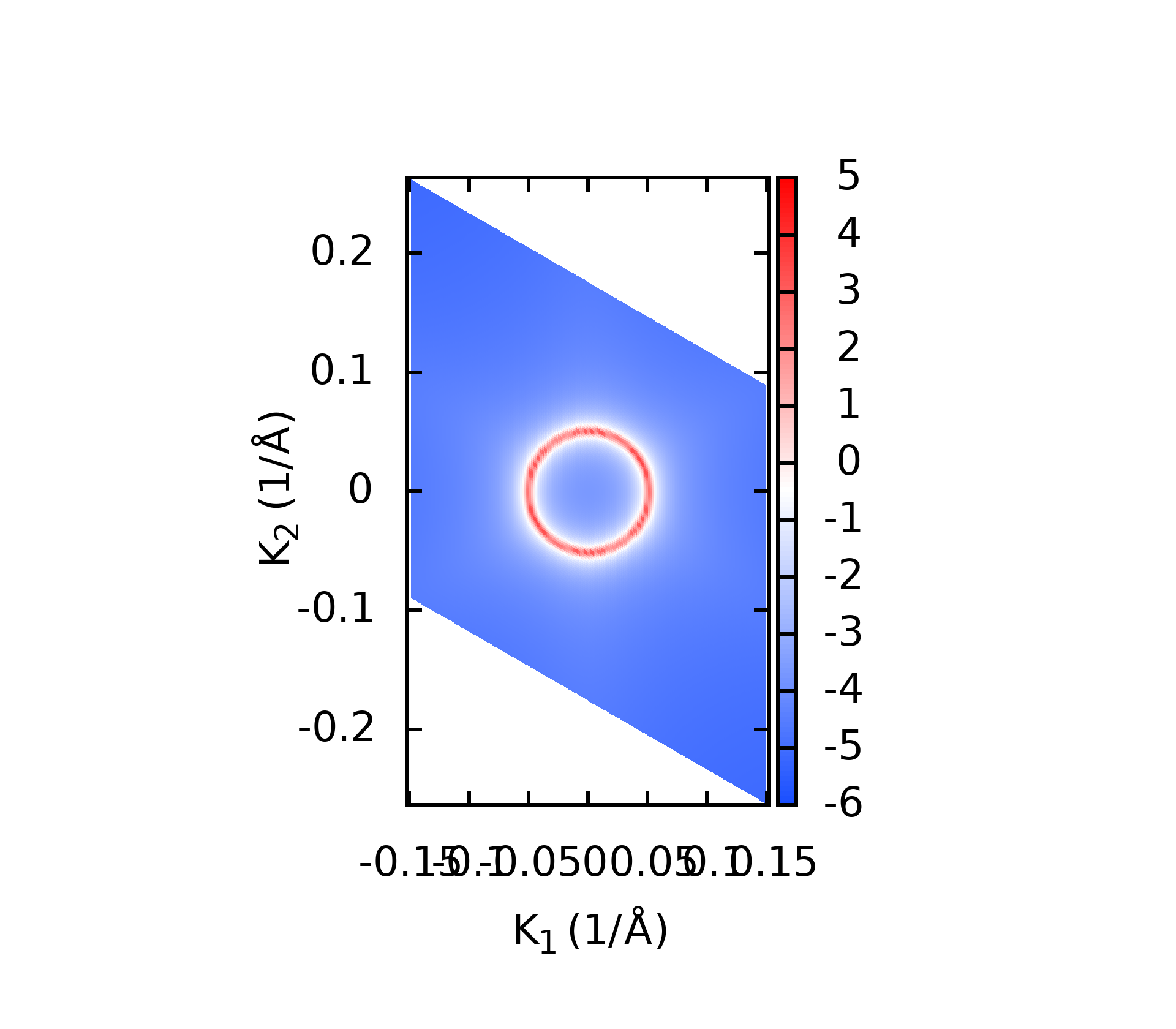
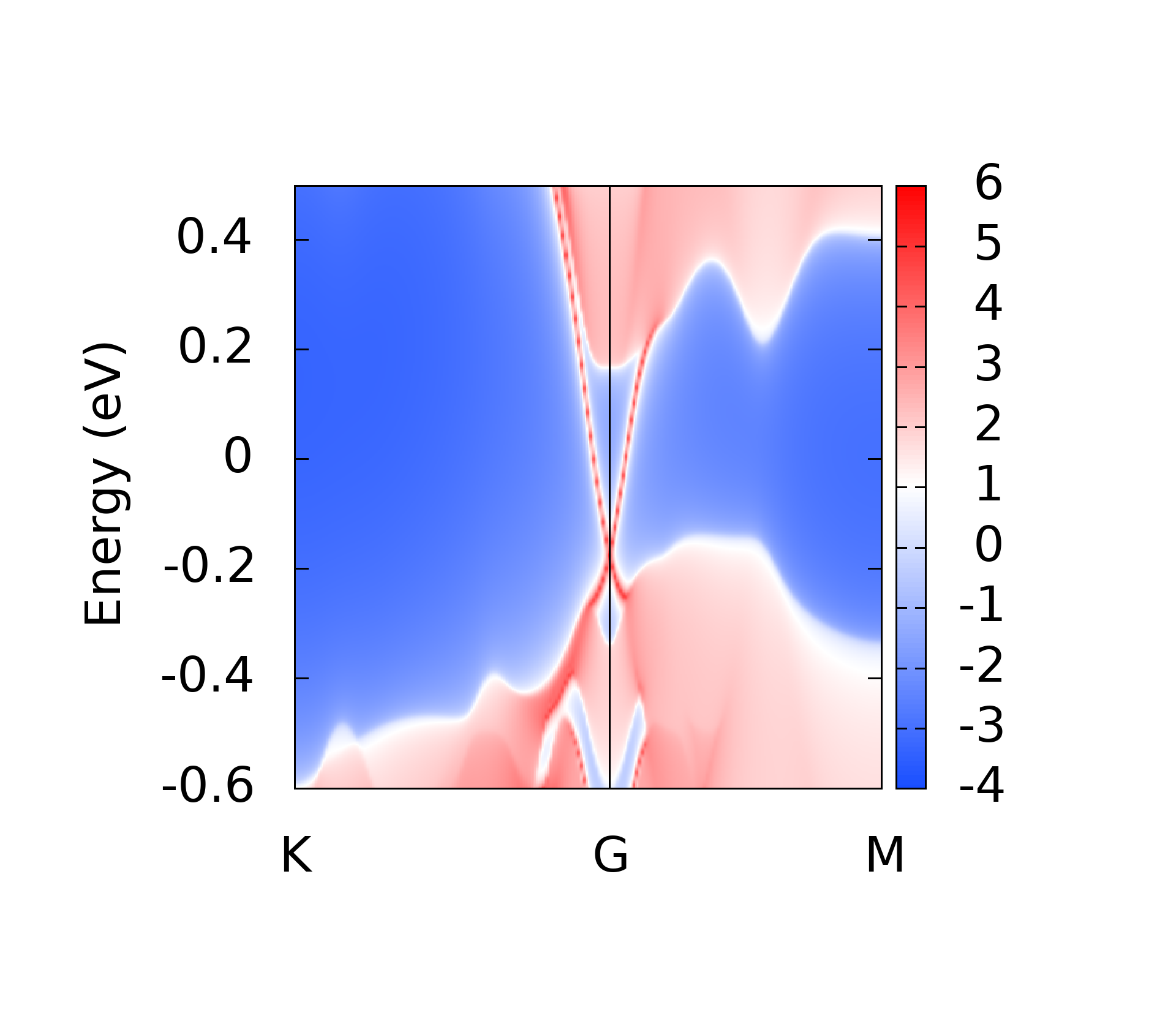
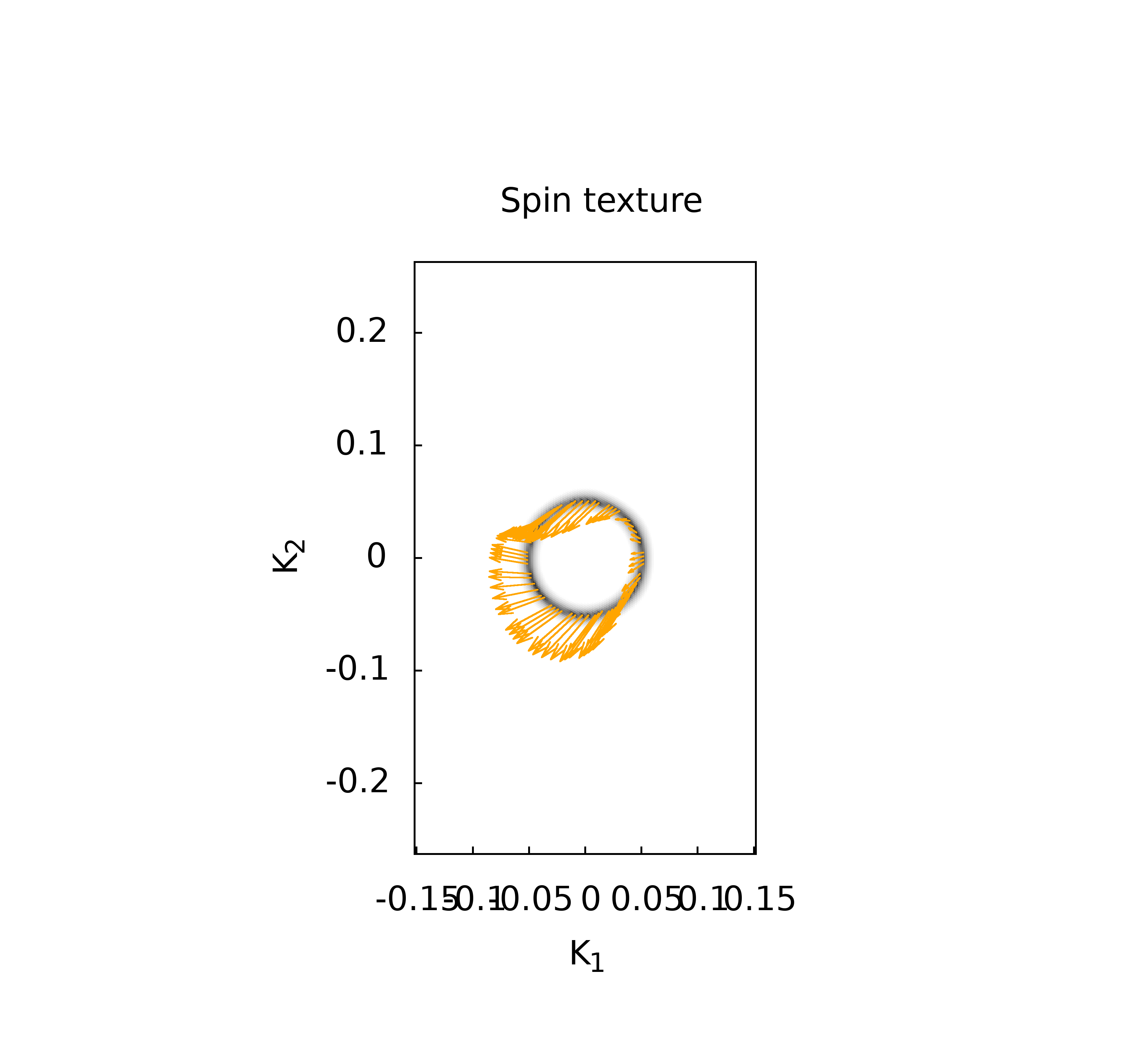
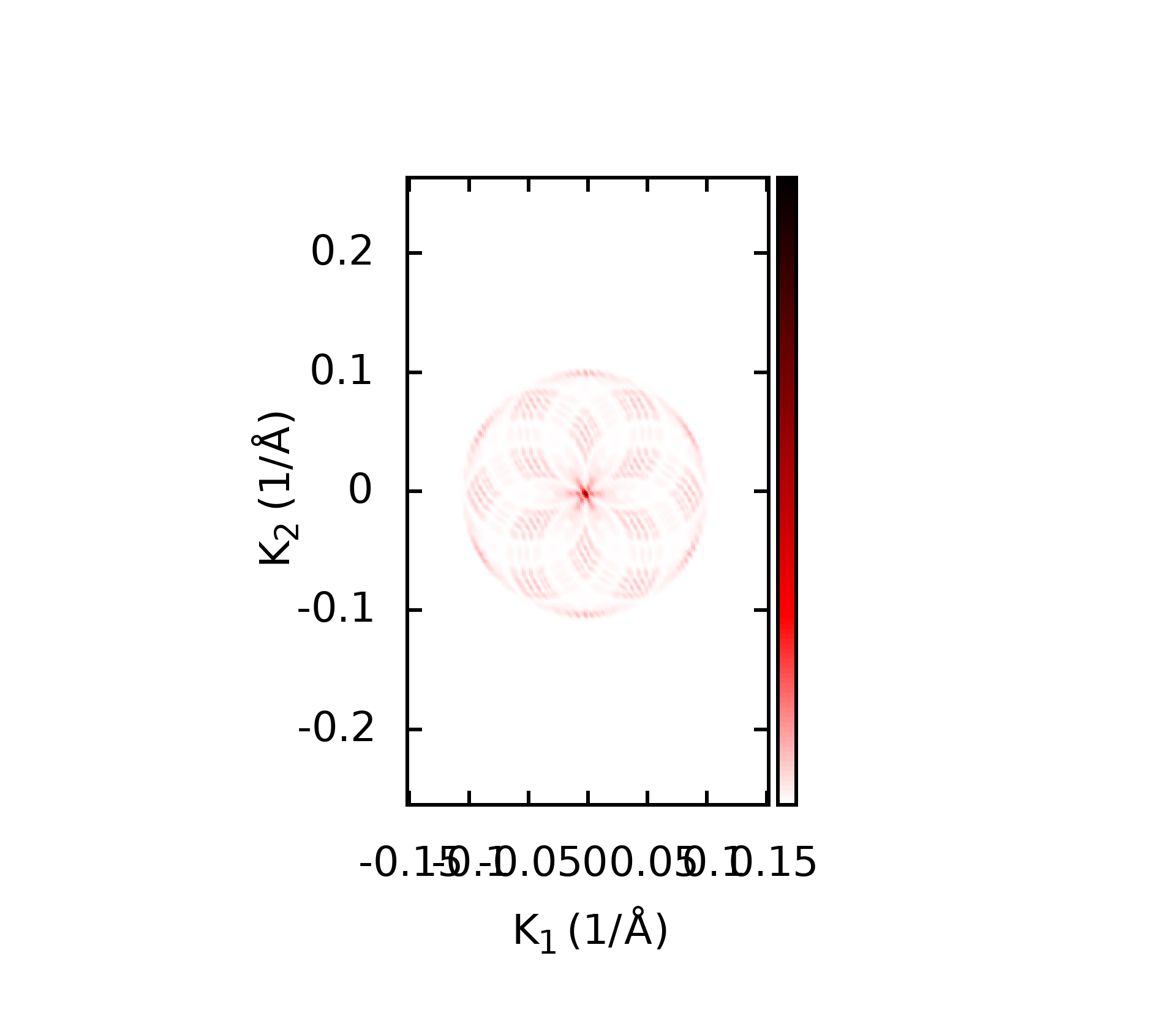
上面的这些计算结果和我在WannierTools中得到的基本一致, 不过还有一些问题, 我正在学习wannier90以及WannierTools相关的内容, 这些不一致的结果我也将会找到原因慢慢修正.
{:.warning}
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。
{:.info}

|
yxliphy@gmail.com |
 wechat
wechat alipay
alipay