学习第一性计算也有一段时间了,这里利用VASP+Wannier90+WannierTools来完整复现一下$Bi_2Se_3$这个材料的一些拓扑性质.
先整理下大致的流程,首先通过VASP自洽计算,得到收敛结果. 第二步结合Wannier90得到Tight binding模型的数据信息. 第三步就是在第二步的基础上得到利用wannier90_hr.dat来计算拓扑相关的性质.
VASP
VASP的计算中需要分两步进行, 首先不考虑SOC进行一次电子自洽, 这里我是直接使用WannierTools文档中$Bi_2Se_3$的晶体结构来计算的, 所以并没有进行结构优化方面的计算.
No-SOC
VASP计算需要的文件内容如下
- INCAR
SYSTEM = NaCl
ICHARG = 2
ISTART = 0
ISYM = 0
ISPIN = 2 # 这里需要考虑自旋, 但是此时并没有打开自旋轨道耦合
GGA = PE
# MAGMOM = 6*0 2*4 2*0
PREC = Normal
ENCUT = 500
ALGO = FAST
EDIFF = 1E-4
EDIFFG = -0.02
LREAL = Auto
ISIF =0
IVDW = 11
NELM = 500
NELMIN = 5
NSW = 0
IBRION = -1
ISMEAR = 0
SIGMA = 0.05
NWRITE = 2
LWAVE = .T.
LCHARG = .T.
LORBIT = 11
##SOC##
# LSORBIT = .TRUE.
# SAXIS = 0 0 1
# NBANDS = 128
# LMAXMIX = 4
# GGA_COMPAT = .FALSE
- POSCAR
Bi2Se3 1.0 -2.069 -3.583614 0.000000 2.069 -3.583614 0.000000 0.000 2.389075 9.546667 Bi Se 2 3 Direct 0.3990 0.3990 0.6970 0.6010 0.6010 0.3030 0 0 0.5 0.2060 0.2060 0.1180 0.7940 0.7940 0.8820 - KPOINTS
Monkhorst Pack 0 G 12 12 3 0 0 0 - POTCAR
至于赝势文件, 自行准备, 没有什么需要强调的.
准备好文件之后, 开始计算即可, 计算收敛之后会得到CHGCAR文件, 这个文件用来进行打开SOC时的收敛计算.
OPEN-SOC
新建一个文件夹, 将上一步计算得到的CHGCAR复制过来, 并将原来的INCAR进行修改
- INCAR
SYSTEM = NaCl
ICHARG = 11 # 利用上一步自洽得到的CHGCAR计算
ISTART = 0
ISYM = 0
ISPIN = 2 # 这里需要考虑自旋, 但是此时并没有打开自旋轨道耦合
GGA = PE
# MAGMOM = 6*0 2*4 2*0
PREC = Normal
ENCUT = 500
ALGO = FAST
EDIFF = 1E-4
EDIFFG = -0.02
LREAL = Auto
ISIF =0
IVDW = 11
NELM = 500
NELMIN = 5
NSW = 0
IBRION = -1
ISMEAR = 0
SIGMA = 0.05
NWRITE = 2
LWAVE = .T.
LCHARG = .T.
LORBIT = 11
##SOC##
LSORBIT = .TRUE. # 打开自旋轨道耦合
SAXIS = 0 0 1 # 确定自旋极化方向
# NBANDS = 128
LMAXMIX = 4
GGA_COMPAT = .FALSE
其余的三个输入文件都不需要改动, 然后进行自洽收敛.
Wannier90
接下来就是利用Wannier90来进行关于Tight Binding相关的计算, 还是在No-SOC自洽计算的基础上, 新建文件夹复制其CHGCAR文件, 修改INCAR如下
- INCAR
SYSTEM = NaCl
ICHARG = 11 # 利用上一步自洽得到的CHGCAR计算
ISTART = 0
ISYM = 0
ISPIN = 2 # 这里需要考虑自旋, 但是此时并没有打开自旋轨道耦合
GGA = PE
# MAGMOM = 6*0 2*4 2*0
PREC = Normal
ENCUT = 500
ALGO = FAST
EDIFF = 1E-4
EDIFFG = -0.02
LREAL = Auto
ISIF =0
IVDW = 11
NELM = 500
NELMIN = 5
NSW = 0
IBRION = -1
ISMEAR = 0
SIGMA = 0.05
NWRITE = 2
LWAVE = .T.
LCHARG = .T.
LORBIT = 11
##SOC##
LSORBIT = .TRUE. # 打开自旋轨道耦合
SAXIS = 0 0 1 # 确定自旋极化方向
# NBANDS = 128
LMAXMIX = 4
GGA_COMPAT = .FALSE
# Wannier90
LWANNIER90 = .TRUE. # 打开wannier90的计算
然后设置wannier90.win文件来控制投影计算
num_wann = 30 # 设置需要投影的Wannier轨道
num_bands = 64 # 这个值右前一步的自洽计算得到
dis_num_iter=1000
num_iter=0
iprint=2
dis_win_min = -2.0
dis_win_max = 18.0
dis_froz_min = -2.0000
dis_froz_max = 5.5000
!hr_plot =.true.
write_hr=.true.
begin projections # 设置原子的投影轨道
Bi : px; py; pz
Se : px; py; pz
end projections
!use_bloch_phases = T
spinors = .true. # 考虑自旋
begin unit_cell_cart
-2.0690000 -3.5836140 0.0000000
2.0690000 -3.5836140 0.0000000
0.0000000 2.3890750 9.5466670
end unit_cell_cart
begin atoms_cart
Bi 0.0000000 -1.1945387 6.6540269
Bi 0.0000000 -3.5836143 2.8926401
Se 0.0000000 1.1945375 4.7733335
Se 0.0000000 -1.1945381 1.1265067
Se 0.0000000 -3.5836149 8.4201603
end atoms_cart
这里需要说明一下num_wann这个参数的设置,首先可以知道元胞内共有5个原子, 而且每个原子都需要由三个轨道投影, 所以一共有$3\times 5=15$个轨道, 但是此时需要考虑自旋, 则共有$3\times 5\times2=30$个轨道需要投影.
准备好INCAR,POSCAR,POTCAR,KPOINTS,wannier90.win着四个文件之后, 就可以开始计算了. 相比于前面只需要修改INCAR文件中的内容即可. 完成计算之后可以得到wannier90.amn,wannier90.chk,wannier90.eig,wannier90.mmn,wannier90.wout,wannier90_wsvec.dat这些文件.
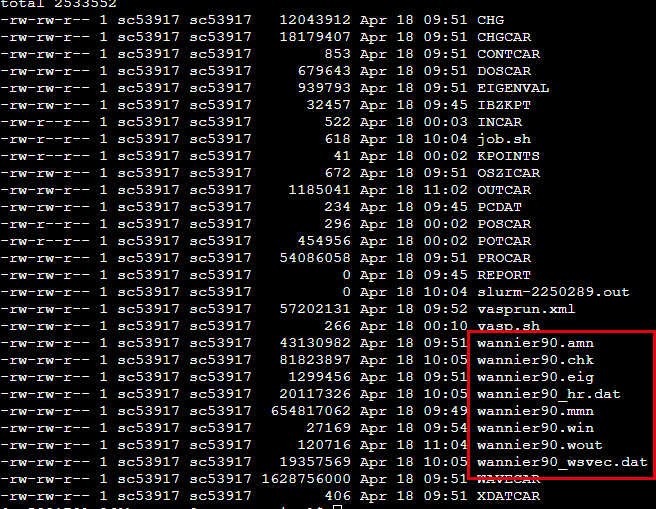
得到这些文件之后,开始利用Wannier90计算Tight Bind所需要的的数据,执行
wannier90.x wannier90
可以最终得到wannier90_hr.dat这个文件,到此Wannier90的计算结束, 之后就可以利用这个数据来进行拓扑方面的计算.
WannierTools
下面进行拓扑方面的计算,将计算得到的wannier90_hr.dat单独复制到一个文件夹, 设置WannierTools所需要的的wt.in
&TB_FILE
Hrfile = 'wannier90_hr.dat'
Package = 'VASP' ! obtained from VASP, it could be 'VASP', 'QE', 'Wien2k', 'OpenMx'
/
LATTICE
Angstrom
-2.069 -3.583614 0.000000 ! crystal lattice information
2.069 -3.583614 0.000000
0.000 2.389075 9.546667
ATOM_POSITIONS
5 ! number of atoms for projectors
Direct ! Direct or Cartisen coordinate
Bi 0.3990 0.3990 0.6970
Bi 0.6010 0.6010 0.3030
Se 0.0000 0.0000 0.5000
Se 0.2060 0.2060 0.1180
Se 0.7940 0.7940 0.8820
PROJECTORS
3 3 3 3 3 ! number of projectors
Bi px py pz ! projectors
Bi px py pz
Se px py pz
Se px py pz
Se px py pz
SURFACE ! Specify surface with two vectors, see doc
1 0 0
0 1 0
!> bulk band structure calculation flag
&CONTROL
BulkBand_calc = T
BulkBand_points_calc = T
DOS_calc = T
SlabBand_calc = T
SlabBandWaveFunc_calc = T
SlabBand_plane_calc = T
WireBand_calc = T
SlabSS_calc = T
SlabArc_calc = T
SlabQPI_calc = T
Z2_3D_calc = T
SlabSpintexture_calc = T
Wanniercenter_calc = T
/
&SYSTEM
NSLAB = 4 ! for thin film system
NSLAB1= 2 ! nanowire system
NSLAB2= 2 ! nanowire system
NumOccupied = 15 ! NumOccupied
SOC = 1 ! soc
E_FERMI = 4.9687 ! e-fermi, a global shift of the energy levels
surf_onsite= 0.0 ! surf_onsite
/
&PARAMETERS
Eta_Arc = 0.001 ! infinite small value, like brodening
E_arc = 0.0 ! energy level for contour plot of spectrum
OmegaNum = 400 ! omega number
OmegaMin = -0.6 ! energy interval
OmegaMax = 0.5 ! energy interval
Nk1 = 101 ! number k points odd number would be better
Nk2 = 101 ! number k points odd number would be better
Nk3 = 101 ! number k points odd number would be better
NP = 5 ! number of principle layers
Gap_threshold = 0.01 ! threshold for FindNodes_calc output
/
KPATH_BULK ! k point path
4 ! number of k line only for bulk band
G 0.00000 0.00000 0.0000 Z 0.00000 0.00000 0.5000
Z 0.00000 0.00000 0.5000 F 0.50000 0.50000 0.0000
F 0.50000 0.50000 0.0000 G 0.00000 0.00000 0.0000
G 0.00000 0.00000 0.0000 L 0.50000 0.00000 0.0000
KPATH_SLAB
2 ! numker of k line for 2D case
K 0.33 0.67 G 0.0 0.0 ! k path for 2D case
G 0.0 0.0 M 0.5 0.5
KPLANE_SLAB
-0.1 -0.1 ! Original point for 2D k plane
0.2 0.0 ! The first vector to define 2D k plane
0.0 0.2 ! The second vector to define 2D k plane for arc plots
KPLANE_BULK
0.00 0.00 0.50 ! Original point for 3D k plane
1.00 0.00 0.00 ! The first vector to define 3d k space plane
0.00 0.50 0.00 ! The second vector to define 3d k space plane
KCUBE_BULK
-0.50 -0.50 -0.50 ! Original point for 3D k plane
1.00 0.00 0.00 ! The first vector to define 3d k space plane
0.00 1.00 0.00 ! The second vector to define 3d k space plane
0.00 0.00 1.00 ! The third vector to define 3d k cube
WANNIER_CENTRES ! copy from wannier90.wout
Cartesian
0.000001 -1.194434 6.617259
0.000041 -1.194660 6.617233
-0.000003 -1.188083 6.631973
0.000159 -1.188134 6.632086
0.000080 -1.200875 6.632263
-0.000066 -1.200732 6.632358
0.000027 -3.583694 2.929604
-0.000171 -3.583517 2.929473
0.000082 -3.590239 2.914729
0.000047 -3.589978 2.914565
-0.000105 -3.577213 2.914447
0.000111 -3.577363 2.914243
0.000003 1.194539 4.773298
0.000006 1.194535 4.773338
0.000042 1.194556 4.773340
-0.000034 1.194539 4.773342
-0.000012 1.194561 4.773333
-0.000007 1.194524 4.773332
0.000026 -1.194484 1.121831
-0.000013 -1.194453 1.121916
-0.000005 -1.212092 1.141185
0.000003 -1.212120 1.141212
0.000051 -1.177254 1.142222
0.000009 -1.177300 1.142218
-0.000003 -3.583717 8.424777
-0.000007 -3.583669 8.424788
-0.000030 -3.566052 8.405460
-0.000054 -3.566017 8.405465
-0.000042 -3.600902 8.404447
0.000034 -3.600867 8.404449
0.000001 -1.194434 6.617259
0.000041 -1.194660 6.617233
-0.000003 -1.188083 6.631973
0.000159 -1.188134 6.632086
0.000080 -1.200875 6.632263
-0.000066 -1.200732 6.632358
0.000027 -3.583694 2.929604
-0.000171 -3.583517 2.929473
0.000082 -3.590239 2.914729
0.000047 -3.589978 2.914565
-0.000105 -3.577213 2.914447
0.000111 -3.577363 2.914243
0.000003 1.194539 4.773298
0.000006 1.194535 4.773338
0.000042 1.194556 4.773340
-0.000034 1.194539 4.773342
-0.000012 1.194561 4.773333
-0.000007 1.194524 4.773332
0.000026 -1.194484 1.121831
-0.000013 -1.194453 1.121916
-0.000005 -1.212092 1.141185
0.000003 -1.212120 1.141212
0.000051 -1.177254 1.142222
0.000009 -1.177300 1.142218
-0.000003 -3.583717 8.424777
-0.000007 -3.583669 8.424788
-0.000030 -3.566052 8.405460
-0.000054 -3.566017 8.405465
-0.000042 -3.600902 8.404447
0.000034 -3.600867 8.404449
上面的设置中WannierCenter的数据可以通过前一步计算得到的wannier90.wout中得到.准备好了wannier90_hr.dat,wt.in这两个文 件之后, 就可以开始拓扑性质方面的计算了,至于如何控制计算,可以自行参考WannierTools的帮助手册, 或者查看我其他相关WannierTools的 计算博客.
这里在进行计算的时候, 需要设置费米能量, 这个值需要我们从自洽结果的OUTCAR文件中寻找
grep E-fermi OUTCAR
得到费米能并设置好之后,执行
wt.x wt.in
开始计算,计算结束后会有一堆.gnu的文件,这是gnuplot绘图的命令文件,这里提供一个脚本,可以批量执行
#!/bin/sh
#============ get the file name ===========
Folder_A=$(pwd)
for file_a in ${Folder_A}/*.gnu
do
gnuplot $file_a
done
代码的本意就是对所有后缀为.gnu的文件, 执行gnuplot命令, 这样最后就可以得到所有的绘图结果了.下面是一些计算的结果
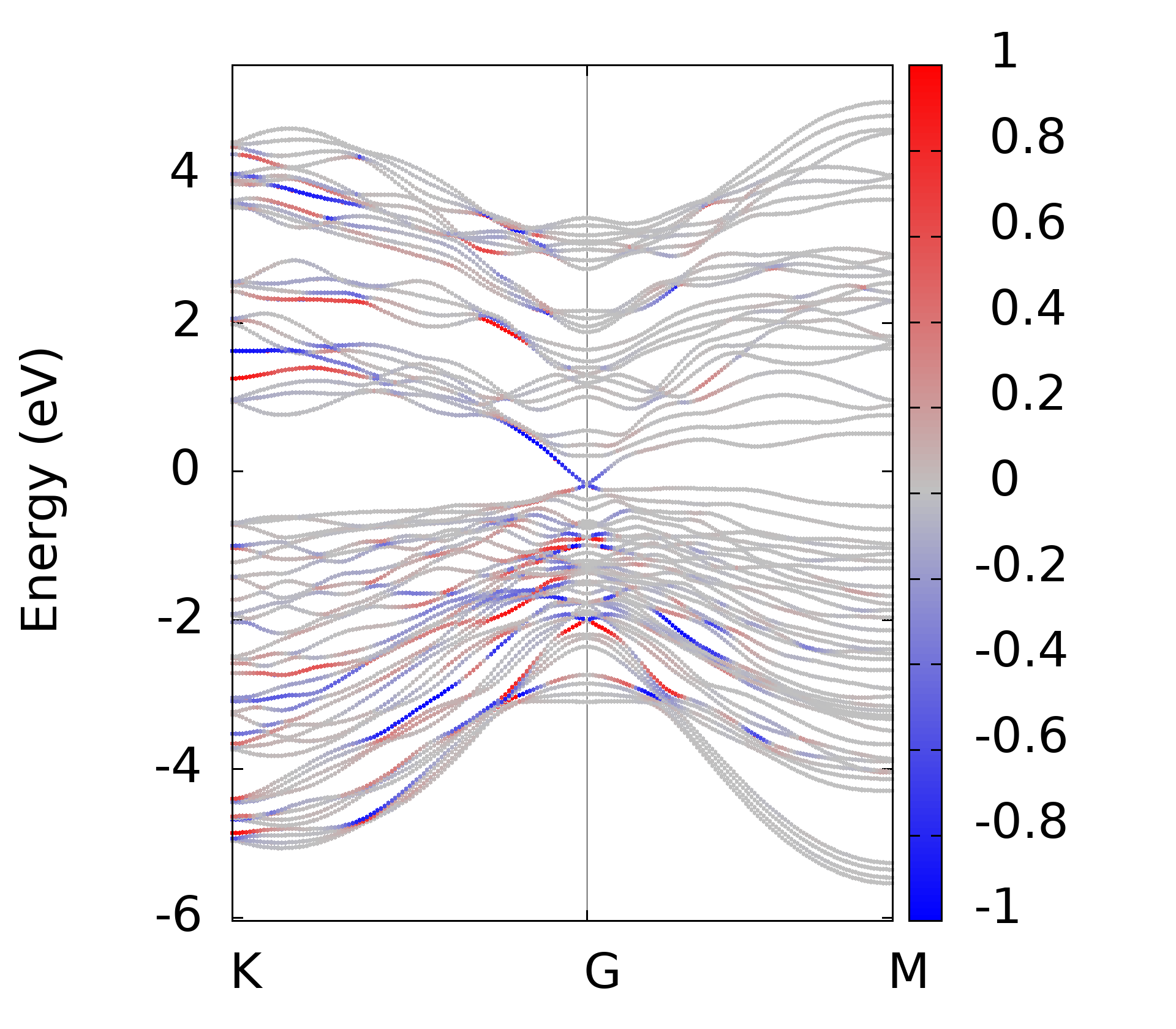
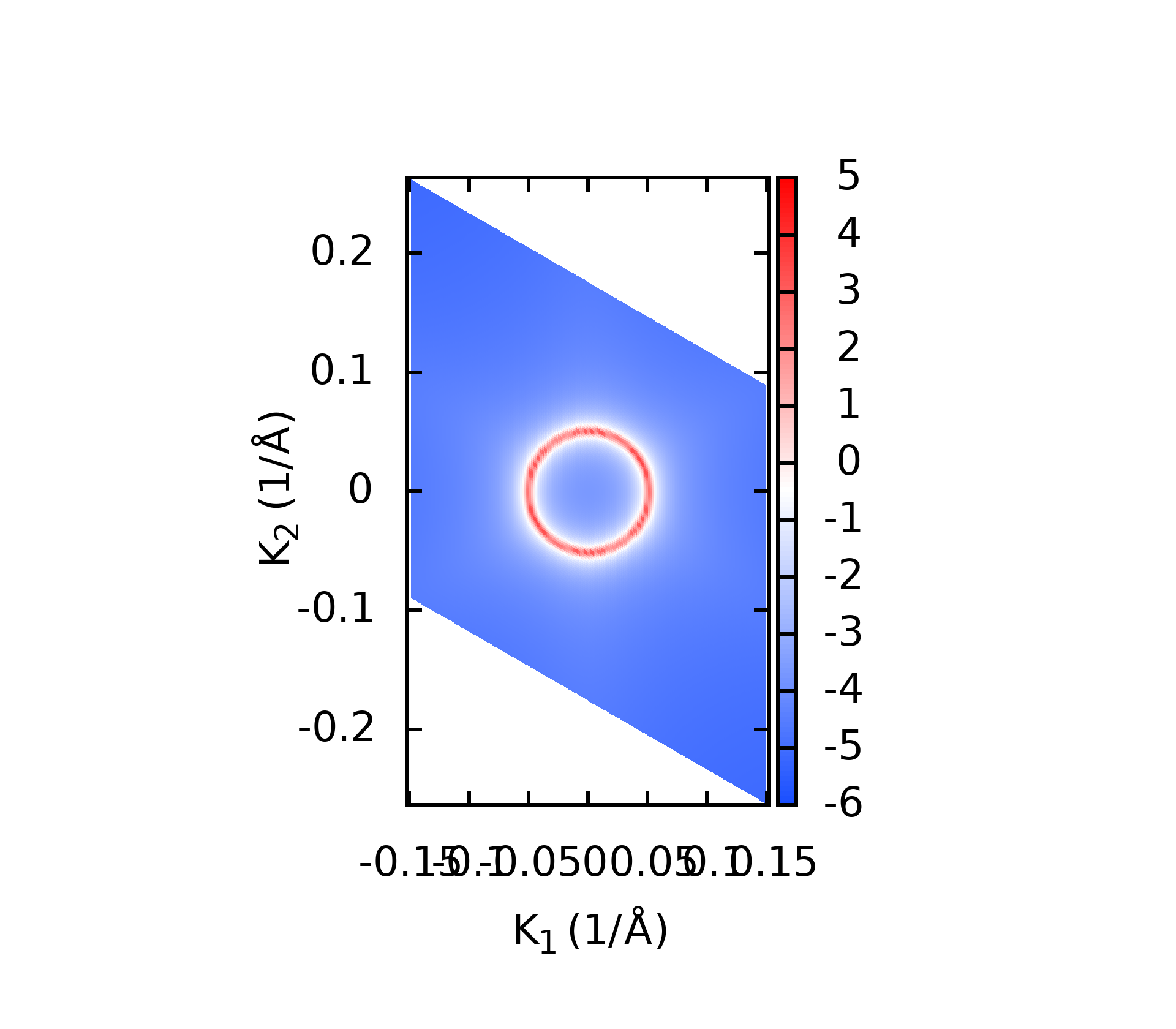
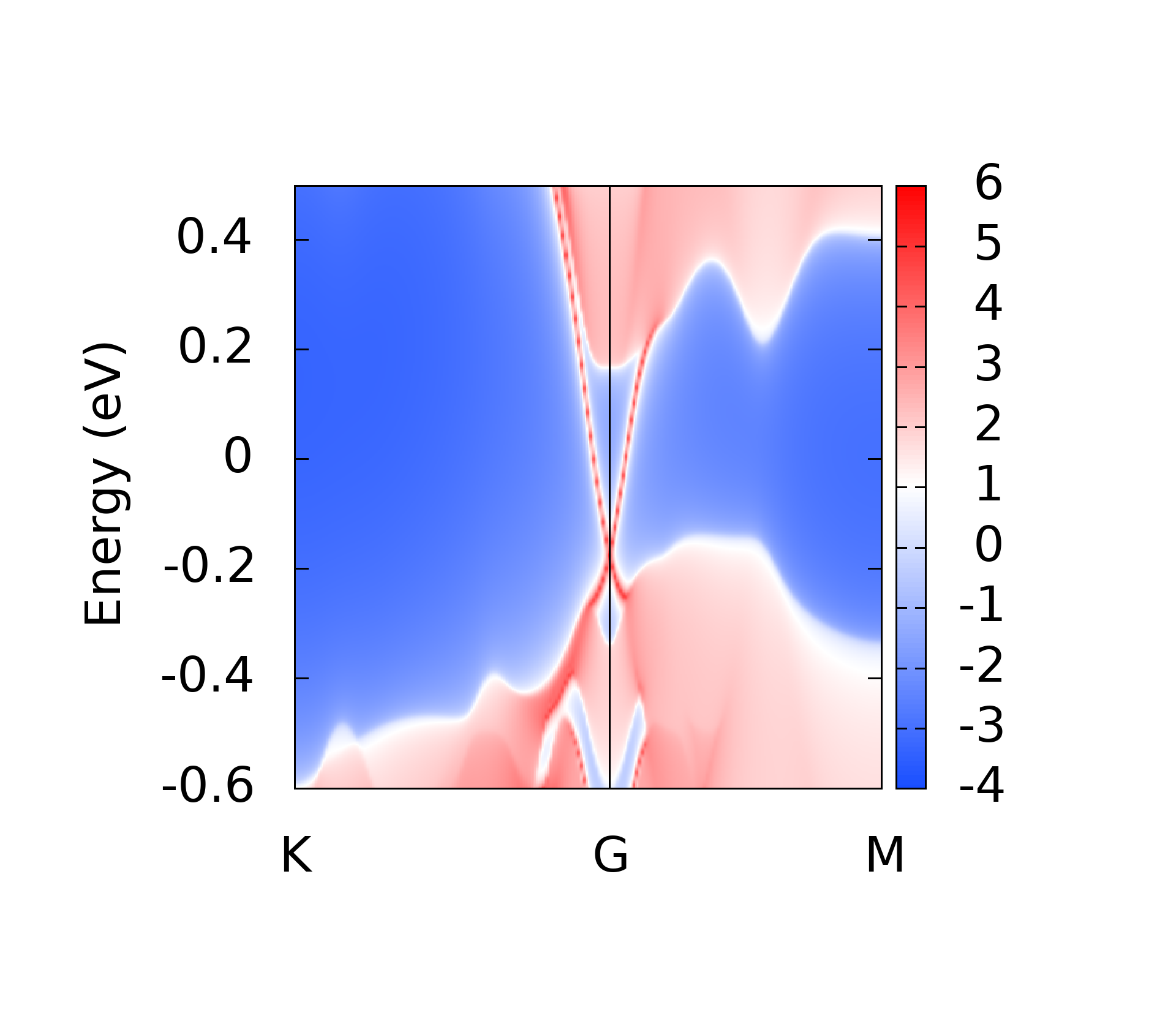
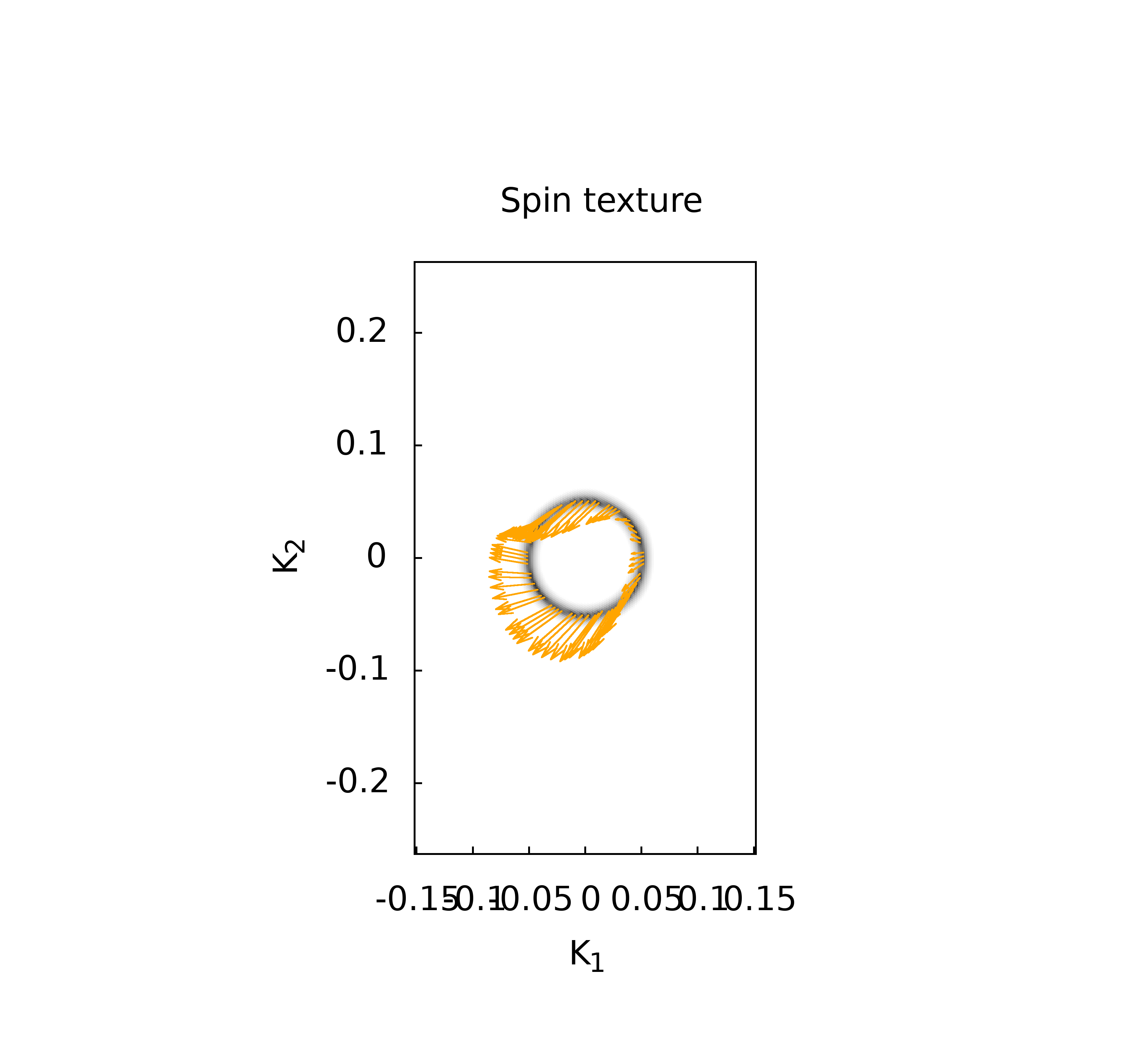
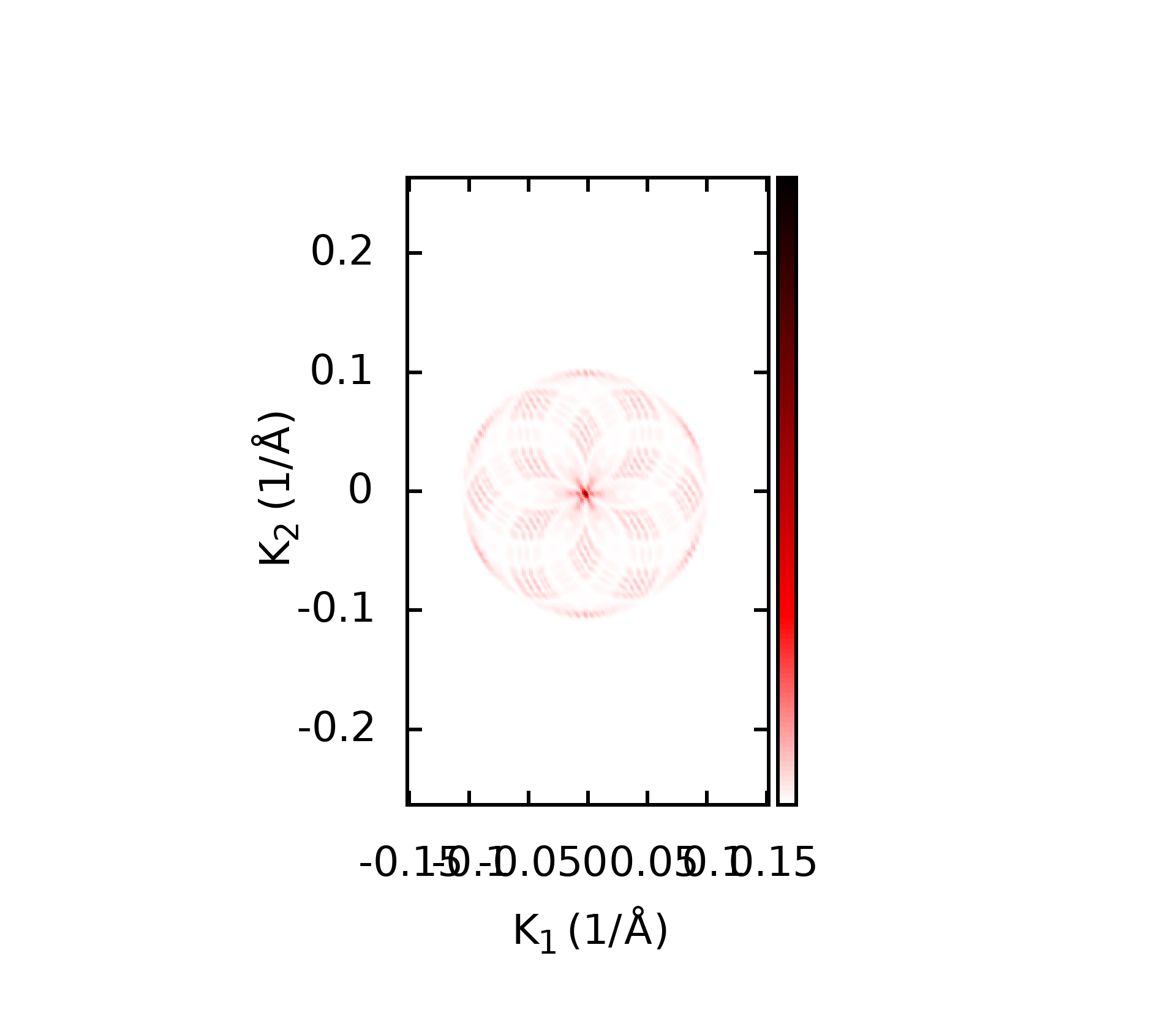
上面的这些计算结果和我在WannierTools中得到的基本一致, 不过还有一些问题, 我正在学习wannier90以及WannierTools相关的内容, 这些不一致的结果我也将会找到原因慢慢修正.
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。

|
yxli406@gmail.com |