这篇博客整理一下怎么利用拓扑量子化学的方法来判断一个材料是拓扑还是半金属还是平庸的。
前言
在之前的学习中,判断一个材料是否为拓扑的,主要还是通过是否存在表面态以及拓扑不变量进行研究的,当然还有新的方案,比如拓扑量子化学的方法,通过能带在高对称 点的不可约表示来直接判断是否为拓扑的,这里就想利用这个方法来研究Bi$_2$Se$_3$的拓扑性质。
利用VASP计算高对称点的能带不可约表示主要有下面的一些步骤
- 自洽计算的到收敛的
CHGCAG - 利用自洽的到的
CHGCAG在高对称点计算波函数WAVECAR - 利用高对称点的
WAVECAR结合irvsp来得到高对称点对称操作的本征值信息 - 利用
vasp2trace来得到trace.txt文件,上传到BCS网站上面来判断拓扑与否不考虑SOC
首先进行自洽计算
自洽计算
- POSCAR
http://materials.springer.com/isp/crystallographic/docs/sd_0541504
4.138 4.138 28.640
.28867513459481288225 -0.50000 0.333333333
.28867513459481288225 0.50000 0.333333333
-.57735026918962576450 0.00000 0.333333333
Bi Se
2 3
Cart
0.0 0.0 0.399
0.0 0.0 -0.399
0.0 0.0 0.206
0.0 0.0 -0.206
0.0 0.0 0.0
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
- KPOINTS
pymatgen 4.7.6+ generated KPOINTS with grid density = 406 / atom
0
Gamma
10 10 10
- INCAR
SYSTEM = Bi2Se3
ISTART = 0
ICHARG = 2
IALGO = 38
EDIFF = 1E-7
#EDIFFG = 1E-6
LREAL = False
ENCUT = 500
#NBANDS= 70
ISMEAR = -5
SIGMA = 0.05
#spin orbit coupling
#LSORBIT =.TRUE.
#SAXIS = 0 0 1
#ISPIN = 2
#MAGMOM = 100*0.0
LORBIT = 11
LWAVE = .FALSE.
LMAXMIX = 4
NPAR = 4
高对称点波函数计算
- POSCAR
http://materials.springer.com/isp/crystallographic/docs/sd_0541504
4.138 4.138 28.640
.28867513459481288225 -0.50000 0.333333333
.28867513459481288225 0.50000 0.333333333
-.57735026918962576450 0.00000 0.333333333
Bi Se
2 3
Cart
0.0 0.0 0.399
0.0 0.0 -0.399
0.0 0.0 0.206
0.0 0.0 -0.206
0.0 0.0 0.0
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
- INCAR
SYSTEM = Bi2Se3
ISTART = 0
ICHARG = 11
#LCHARG = .FALSE.
IALGO = 38
EDIFF = 1E-7
#EDIFFG = 1E-6
LREAL = False
ENCUT = 500
#NBANDS= 70
ISMEAR = 0
SIGMA = 0.05
#spin orbit coupling
#LSORBIT =.TRUE.
#SAXIS = 0 0 1
#ISPIN = 2
#MAGMOM = 100*0.0
LORBIT = 11
#LWAVE = .FALSE.
LMAXMIX = 4
#NPAR = 4
- KPOINTS
k-points
4
rec
0.00000000 0.00000000 0.00000000 1.0
0.50000000 0.50000000 0.50000000 1.0
0.50000000 0.50000000 0.00000000 1.0
0.00000000 0.50000000 0.00000000 1.0
这里关于高对称点的信息,可以在irvsp中文件夹中寻找
提交VASP任务之后,就可以得到在高对称点上的波函数文件WAVECAR
irvsp计算
计算高对称点对称操作本征值
在高对称点上的波函数文件WAVECAR之后,执行irvsp来得到结果对称操作的本征值
irvsp -sg 166 >outir &
计算trace文件
vasp2trace
执行vasp2trace之后就可以得到trace.txt文件,将其上传到拓扑量子化学的网站,如下图所示
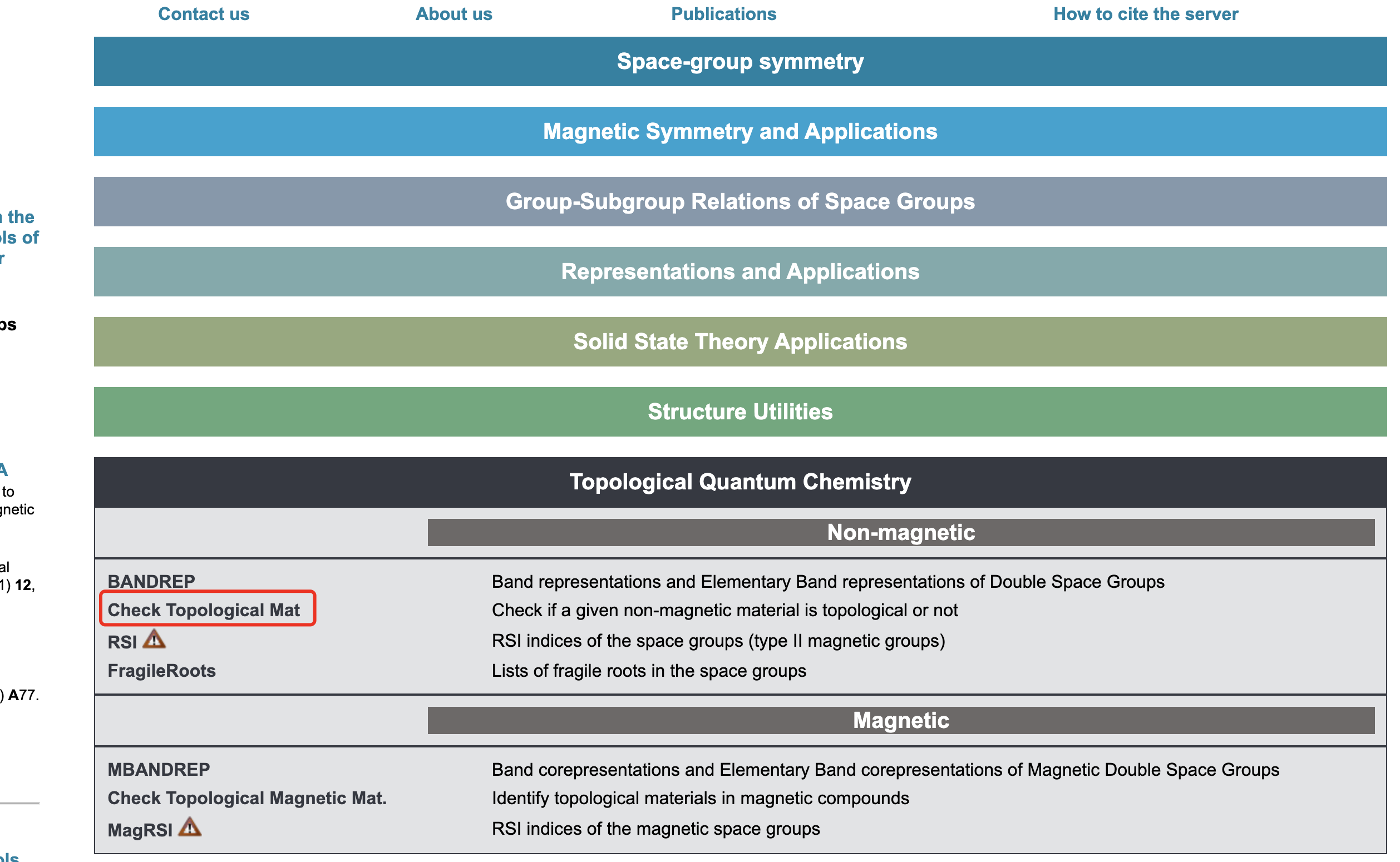
就可以通过拓扑量子化学方法得到的结果

通过上面的结果可以看到,在不考虑SOC的时候,Bi$_2$Se$_3$是平庸的,The set of bands below the Fermi level can be expressed as a Linear Combination of Elementary Band Representations (LCEBR). The compound is a TRIVIAL insulator or topological with trivial symmetry indicators.
考虑SOC
在考虑SOC的时候,计算过程和不考虑SOC时候的情况完全一样,只不过需要修改一下VASP计算时候的文件
自洽计算
- POSCAR
http://materials.springer.com/isp/crystallographic/docs/sd_0541504
4.138 4.138 28.640
.28867513459481288225 -0.50000 0.333333333
.28867513459481288225 0.50000 0.333333333
-.57735026918962576450 0.00000 0.333333333
Bi Se
2 3
Cart
0.0 0.0 0.399
0.0 0.0 -0.399
0.0 0.0 0.206
0.0 0.0 -0.206
0.0 0.0 0.0
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
- INCAR
SYSTEM = Bi2Se3
ISTART = 0
ICHARG = 2
IALGO = 38
EDIFF = 1E-7
#EDIFFG = 1E-6
LREAL = False
ENCUT = 500
#NBANDS= 70
ISMEAR = -5
SIGMA = 0.05
#spin orbit coupling
LSORBIT =.TRUE.
#SAXIS = 0 0 1
#ISPIN = 2
MAGMOM = 100*0.0
LORBIT = 11
LWAVE = .FALSE.
LMAXMIX = 4
NPAR = 4
- KPOINTS
pymatgen 4.7.6+ generated KPOINTS with grid density = 406 / atom
0
Gamma
10 10 10
高对称点波函数计算
- POSCAR
http://materials.springer.com/isp/crystallographic/docs/sd_0541504
4.138 4.138 28.640
.28867513459481288225 -0.50000 0.333333333
.28867513459481288225 0.50000 0.333333333
-.57735026918962576450 0.00000 0.333333333
Bi Se
2 3
Cart
0.0 0.0 0.399
0.0 0.0 -0.399
0.0 0.0 0.206
0.0 0.0 -0.206
0.0 0.0 0.0
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
0.00000000E+00 0.00000000E+00 0.00000000E+00
- INCAR
SYSTEM = Bi2Se3
ISTART = 1
ICHARG =11
IALGO = 38
EDIFF = 1E-7
#EDIFFG = 1E-6
LREAL = False
ENCUT = 500
#NBANDS= 70
ISMEAR = 0
SIGMA = 0.05
#spin orbit coupling
LSORBIT =.TRUE.
#SAXIS = 0 0 1
#ISPIN = 2
MAGMOM = 100*0.0
LORBIT = 11
LWAVE = .TRUE.
LMAXMIX = 4
#NPAR = 4
- KPOINTS
k-points
4
rec
0.00000000 0.00000000 0.00000000 1.0
0.50000000 0.50000000 0.50000000 1.0
0.50000000 0.50000000 0.00000000 1.0
0.00000000 0.50000000 0.00000000 1.0
提交任务之后就可以得到在考虑SOC之后,高对称点上的波函数WAVECAR,接下来就可以使用irvsp来计算了。
irvsp计算
计算高对称点对称操作本征值
在高对称点上的波函数文件WAVECAR之后,执行irvsp来得到结果对称操作的本征值
irvsp -sg 166 >outir &
计算trace文件
vasp2trace
执行vasp2trace之后就可以得到trace.txt文件,将其上传到拓扑量子化学的网站,如下图所示
就可以通过拓扑量子化学方法得到的结果

通过上面的结果可以看到,在考虑SOC的时候,Bi$_2$Se$_3$是拓扑的,The set of bands below the Fermi level cannot be expressed as a Linear Combination of Elementary Band Representations, but it can be expressed as Linear Combination of Elementary Band Representations and disconnected parts of Elementary Band Representations. The compound has been identified as a topological insulator.
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。

|
yxli406@gmail.com |