最近在慢慢接触第一性计算的相关内容,需要补充一些工具,这里就整理记录一下我在学习Wannier90过程中遇到的疑问和自己对其中内容的一些理解.
我在这里完全就先是重复Wannier90给出的例子,在重复的过程中通过文档边学习边理解.
Gallium Arsenide(Example 1)
最主要的就是输入控制文件file.win,这个文件来控制要进行什么样的计算.这里需要说明,要计算不同的性质和物理量,除了控制文件之外还需要其他的数据,这第一个实例中的文件如下

! Gallium Arsenide: Tutorial Example 1
num_wann = 4
num_iter = 20
wannier_plot = true
! SYSTEM
begin unit_cell_cart
bohr
-5.367 0.000 5.367
0.000 5.367 5.367
-5.367 5.367 0.000
end unit_cell_cart
begin atoms_frac
Ga 0.00 0.00 0.00
As 0.25 0.25 0.25
end atoms_frac
begin projections
As:sp3
end projections
! KPOINTS
mp_grid : 2 2 2
begin kpoints
0.0 0.0 0.0
0.0 0.0 0.5
0.0 0.5 0.0
0.0 0.5 0.5
0.5 0.0 0.0
0.5 0.0 0.5
0.5 0.5 0.0
0.5 0.5 0.5
end kpoints
!We set this flag to read the bloch states from
!a formatted file. This is to ensure the example
!works on all platforms. The default (.false.) state
!should be used on production runs
wvfn_formatted=.true.
下面对控制文件中的一些参数进行解释 num_wann:控制需要寻找的Wannier函数的数目.
num_bands:这个参数和file.mmn中的信息相关,表示生成这个文件中能带的数目是多少,就像我在Bi$_2$Se$_3$这个实例中,我可以从vasp的计算中得到计算的能带数目是64.
num_iter:在求解最大局域化Wannier函数的过程中的迭代次数控制,默认值是100.
wannier_plot:用来控制是否计算出费米面相关的信息,最后会产生file.xsf的文件,不过需要用特定的软件来绘图.
begin unit_cell_cart
bohr
-5.367 0.000 5.367
0.000 5.367 5.367
-5.367 5.367 0.000
end unit_cell_cart
这个控制参数用来声明计算体系元胞的信息,unit_cell_cart表示是用直角坐标,这里的bohr表示坐标的单位,也可以设置为Ang,一般默认值是Ang.
begin atoms_frac
Ga 0.00 0.00 0.00
As 0.25 0.25 0.25
end atoms_frac
元胞设置好之后,就需要明确元胞中原子的位置,atoms_frac表明用的是分数坐标来表示原子位置;同样也可以使用atoms_cart,此时就是利用直角坐标表示位置.
begin projections
As:sp3
end projections
每个原子上的投影轨道设置(暂时不太懂,等之后把理论好好研究一番再回来).
mp_grid : 2 2 2
Monkhorst-Pack网格上的撒点密度.
begin kpoints
......................
end kpoints
这个参数在利用vasp结合Wannier90计算的时候,刚开始不会设置,在运行结束之后会自动产生,暂时不明白这个参数的具体含义.
Lead(Example 2)
这个实例中计算文件如下所示
这里的lead.eig中包含的是每个k点处的Block本征值,是为了在这个实例中进行费米面的插值计算准备的.控制输入文件lead.win内容如下
! Lead : Tutorial Example 2
num_wann = 4
num_iter = 20
! SYSTEM
begin unit_cell_cart
bohr
-4.67775 0.00000 4.67775
0.00000 4.67775 4.67775
-4.67775 4.67775 0.00000
end unit_cell_cart
begin atoms_frac
Pb 0.00 0.00 0.00
end atoms_frac
begin projections
Pb:sp3
end projections
! KPOINTS
mp_grid : 4 4 4
begin kpoints
.............................
end kpoints
这里的参数没有过多解释的必要,在前面的实例中都出现过了.在这个控制文件中加入
restart = plot
fermi_energy = 5.2676
fermi_surface_plot = true
之后,重新进行计算,可以得到
所示的文件,这里的lead.bxsf就是插值得到的费米面的信息,只需上面的参数,从名字可就可以理解了.这里的费米能如果是利用vasp进行计算的话,是可以从OUTCAR这个文件中读取的.
grep E-fermi OUTCAR
关于费米能的这个内容,可以参Bi$_2$Se$_3$第一性计算结果重复这篇博客中的内容.
Silicon
文件家中的文件如下图所示

现在仍然只关心输入控制文件silicon.win
num_bands = 12
num_wann = 8
dis_win_max = 17.0d0
dis_froz_max = 6.4d0
dis_num_iter = 120
dis_mix_ratio = 1.d0
num_iter = 50
num_print_cycles = 10
Begin Atoms_Frac
Si -0.25 0.75 -0.25
Si 0.00 0.00 0.00
End Atoms_Frac
Begin Projections
Si : sp3
End Projections
begin kpoint_path
L 0.50000 0.50000 0.5000 G 0.00000 0.00000 0.0000
G 0.00000 0.00000 0.0000 X 0.50000 0.00000 0.5000
X 0.50000 -0.50000 0.0000 K 0.37500 -0.37500 0.0000
K 0.37500 -0.37500 0.0000 G 0.00000 0.00000 0.0000
end kpoint_path
Begin Unit_Cell_Cart
-2.6988 0.0000 2.6988
0.0000 2.6988 2.6988
-2.6988 2.6988 0.0000
End Unit_Cell_Cart
mp_grid = 4 4 4
begin kpoints
......................
End Kpoints
dis_win_max:这个参数是一个能量下的态都被包括进来,用来解纠缠,通常配合使用的还有dis_win_min,它用来设置最低能量范围.
dis_froz_max:这个参数也是用来解纠缠的,暂时不能白这些能量窗口的含义,先学习怎么使用.
dis_mix_ratio:解纠缠时设置的一个混合参数,建议值是[0,1]
begin kpoint_path
L 0.50000 0.50000 0.5000 G 0.00000 0.00000 0.0000
G 0.00000 0.00000 0.0000 X 0.50000 0.00000 0.5000
X 0.50000 -0.50000 0.0000 K 0.37500 -0.37500 0.0000
K 0.37500 -0.37500 0.0000 G 0.00000 0.00000 0.0000
end kpoint_path
用来设置计算能带时候动量空间中的路径.
将上面的输入文件执行完成之后,在加入
restart = plot
bands_plot = true
进行能带的绘制,执行完成之后,得到的文件如下
如果服务器上安装好了gnuplot绘图工具的话,执行
gnuplot silicon_band.gnu
即可绘制能带图.
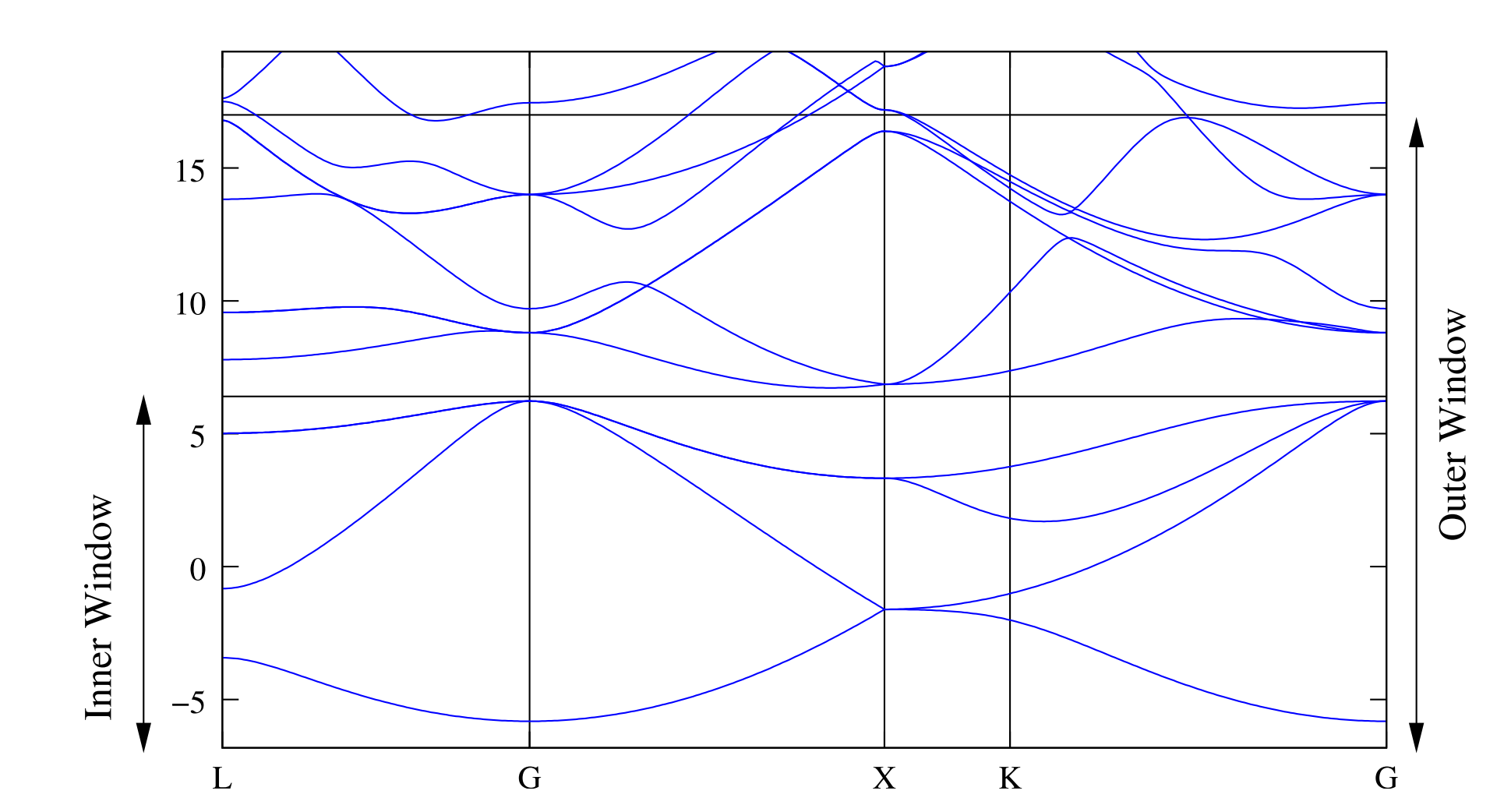
这个能带图中国标级除了两个窗口的位置,与dis_win_max和dis_froz_max有关,其中范围较大的Outer Window就是dis_win_max限定的范围,而范围较小的窗口Inner Window就是dis_froz_max所限定的范围.
Copper
自带的文件如下,里面每个文件到底是如何来的先不关心
先来看看控制文件中的内容
num_bands = 12
num_wann = 7
num_iter = 200
dis_win_max = 38.0
dis_froz_max = 13.0
dis_num_iter = 60
dis_mix_ratio = 1.0d0
! SYSTEM
begin unit_cell_cart
bohr
-3.411 0.000 3.411
0.000 3.411 3.411
-3.411 3.411 0.000
end unit_cell_cart
begin atoms_frac
Cu 0.00 0.00 0.00
end atoms_frac
begin projections
Cu:d
f=0.25,0.25,0.25:s
f=-0.25,-0.25,-0.25:s
end projections
begin kpoint_path
G 0.00 0.00 0.00 X 0.50 0.50 0.00
X 0.50 0.50 0.00 W 0.50 0.75 0.25
W 0.50 0.75 0.25 L 0.00 0.50 0.00
L 0.00 0.50 0.00 G 0.00 0.00 0.00
G 0.00 0.00 0.00 K 0.00 0.50 -0.50
end kpoint_path
! KPOINTS
mp_grid : 4 4 4
begin kpoints
.............
end kpoints
dis_win_max:The upper bound of the outer energy window for the disentanglement procedure. Units are eV.
dis_froz_max:The upper bound of the inner (frozen) energy window for the disentanglement procedure.
dis_num_iter:解纠缠过程中的迭代次数,用来达到最好的关联空间.
dis_mix_ratio:在解纠缠过程中使用这个混合参数来进行收敛,通常1.0是最快的收敛过程.
begin projections
Cu:d
f=0.25,0.25,0.25:s
f=-0.25,-0.25,-0.25:s
end projections
这里的f=0.25,0.25,0.25表示原子中心的坐标,利用的是分数(f)坐标来表示,后面的:s暂时不明白它的含义.
将上面的执行完成之后,正常运行下就会产生copper.wout文件表明执行完成,计算过程中的信息都可以从这个文件中查看.
下面来画费米面,这里的费米能量是12.2103eV,
restart = plot
fermi_energy = 12.2103
fermi_surface_plot = true
绘制能带结构的时候,需要加入动量空间中的路径
begin kpoint_path
G 0.00 0.00 0.00
X 0.50 0.50 0.00
X 0.50 0.50 0.00
W 0.50 0.75 0.25
W 0.50 0.75 0.25
L 0.00 0.50 0.00
L 0.00 0.50 0.00
G 0.00 0.00 0.00
G 0.00 0.00 0.00
K 0.00 0.50 -0.50
end kpoint_path
将前面绘制能带加入的命令替换成
restart = plot
bands_plot = true
结果如下所示
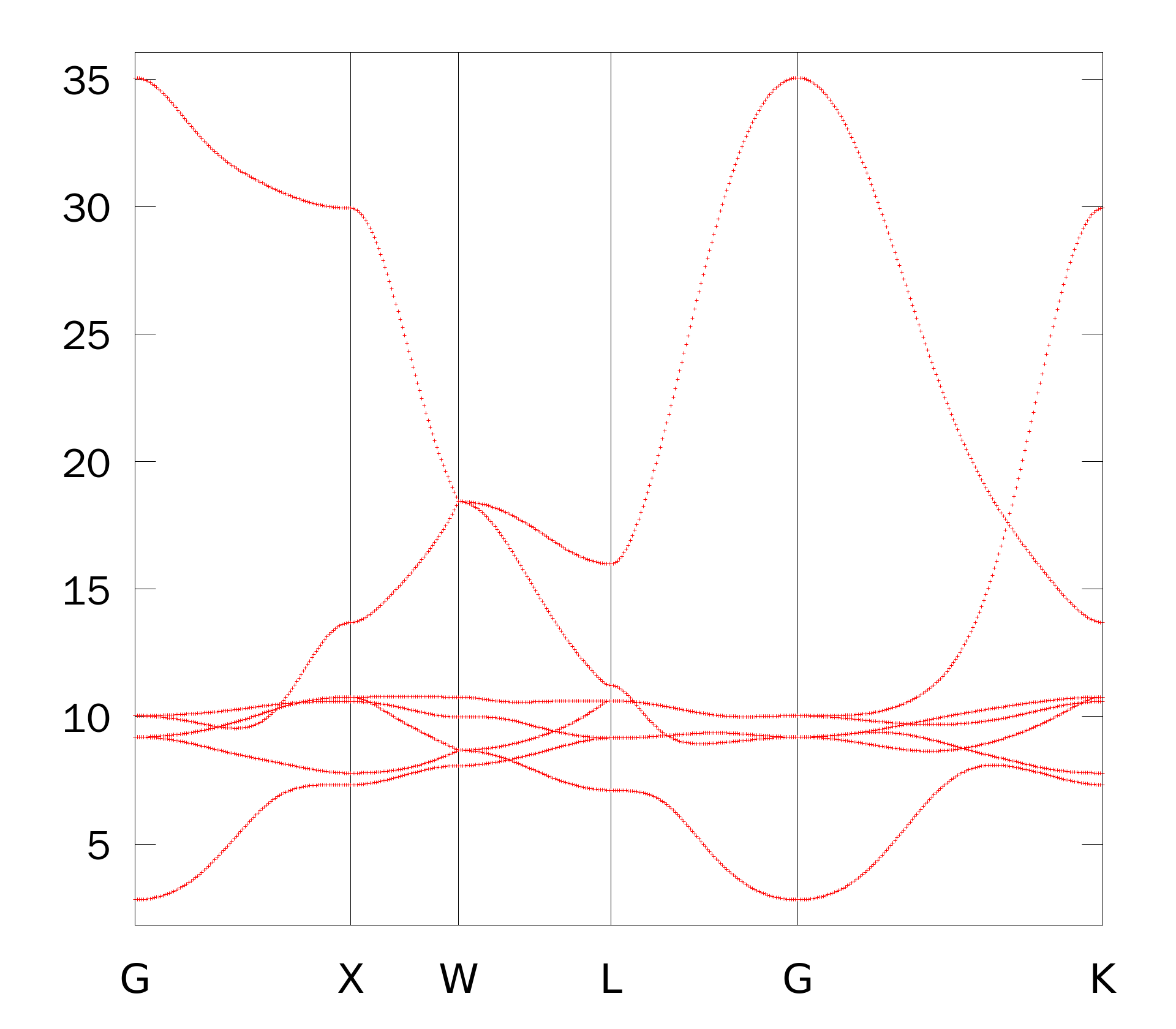
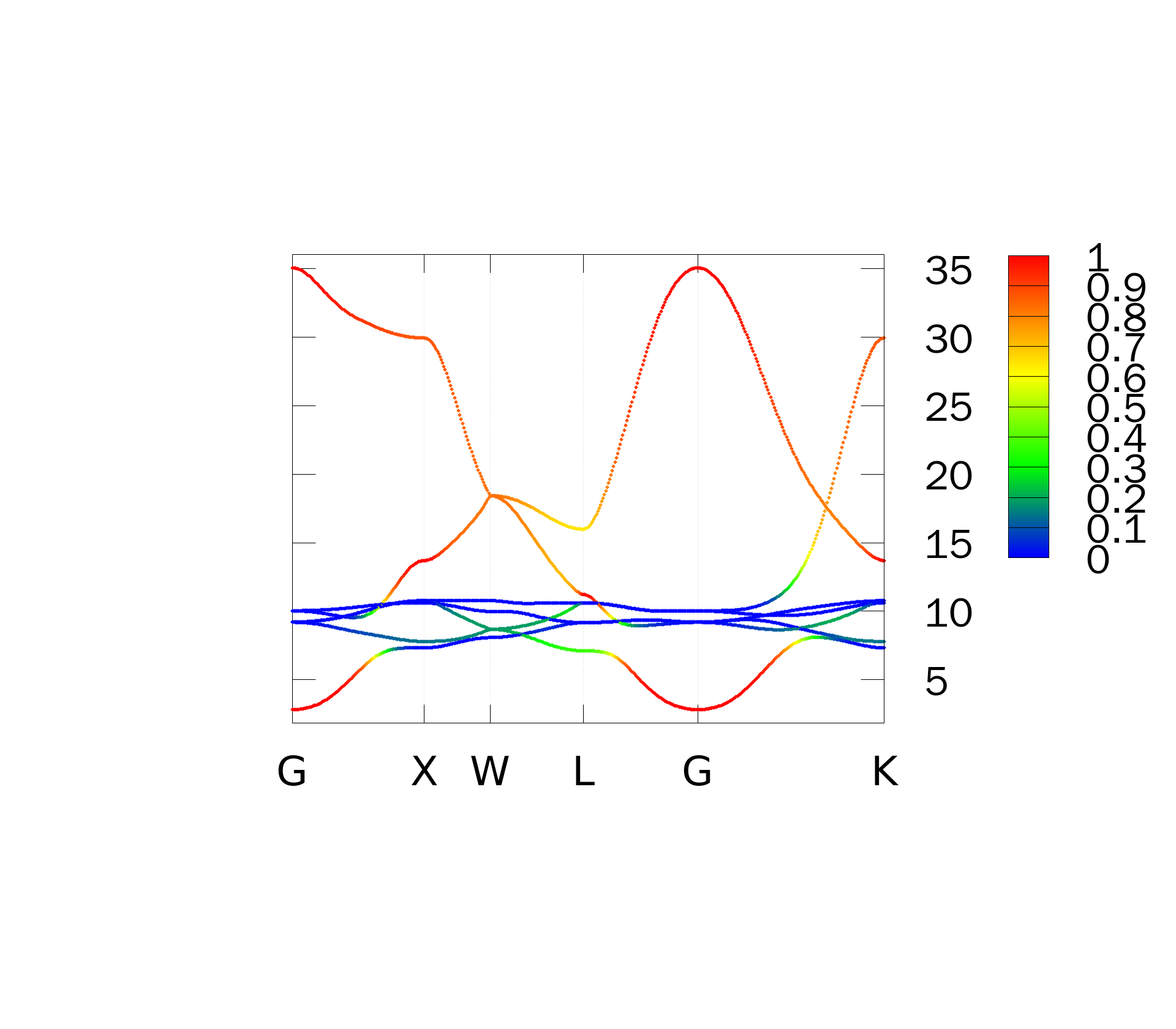
Plot the contribution of the interstitial WF to the bandstructure. Add the following keyword to copper.win
bands_plot_project = 6,7
暂时不是很清楚这个设置的物理含义,猜测是做能带投影的,但是这个数值设置不明白,得到的结果如下
前半部分的学习先告一段路,后面的实例学习需要用到Wannier90的另外一个工具,先要熟悉一下对应的理论知识.
公众号
相关内容均会在公众号进行同步,若对该Blog感兴趣,欢迎关注微信公众号。

|
yxli406@gmail.com |